
Whole-genome characterisation of multidrug resistant monophasic variants of Salmonella Typhimurium from pig production in Thailand
- Published
- Accepted
- Received
- Academic Editor
- Joseph Gillespie
- Subject Areas
- Microbiology, Veterinary Medicine, Epidemiology, Public Health
- Keywords
- Monophasic Salmonella Typhimurium, Pig production, Antimicrobial resistance (AMR), Salmonella pathogenicity islands (SPI), Multidrug resistance (MDR), Whole-genome multilocus sequence typing (wgMLST)
- Copyright
- © 2020 Patchanee et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2020. Whole-genome characterisation of multidrug resistant monophasic variants of Salmonella Typhimurium from pig production in Thailand. PeerJ 8:e9700 https://doi.org/10.7717/peerj.9700
Abstract
Background
Monophasic Salmonella Typhimurium or S. enterica 1,4,[5],12:i:- is among the top five serotypes reported in Thailand. In this study, nineteen monophasic S. Typhimurium from the pig production chain in Chiang Mai and Lamphun provinces during 2011–2014 were sequenced and compared to a globally disseminated clone. Isolates were probed in silico for the presence of antimicrobial resistance genes and Salmonella virulence factors, including Pathogenicity Islands.
Results
All isolates were from sequence type 34 (ST-34) and clustered similarly in core and pangenome genealogies. The two closest related isolates showed differences in only eighteen loci from whole-genome multilocus sequence typing analysis. All 19 isolates carried aminoglycoside and beta-lactam class resistance genes and genes for five or more different antibiotic classes. Seven out of 14 known SPIs were detected, including SPI-5, SPI-13 and SPI-14, which were detected in all isolates.
Conclusions
The multi-drug resistant clone, ST-34 was sampled at all stages of pork production. This clone has infiltrated global agricultural processes and poses a significant public health risk. Differences in the core and accessory genomes of the isolates we collected suggest that strains persist though the pork production process, with evidence of mutation within the core-genome and horizontal acquisition of genes, potentially via sharing of pathogenicity islands and plasmids. This highlights the importance of surveillance and targeted intervention measures to successfully control Salmonella contamination.
Introduction
Salmonella is a globally important cause of diarrhoeal diseases (World Health Organization (WHO), 2015). Annually, there are ninety-million cases with 150,000 deaths of food-borne salmonellosis estimated worldwide (Majowicz et al., 2010). Disease in humans is associated with contaminated or undercooked meats (Bangtrakulnonth et al., 2004; Mossong et al., 2007; Campioni, Moratto Bergamini & Falcao, 2012). Symptoms observed include acute fever, nausea, vomiting and diarrhoea within 8–48 h of infection (Jawetz, Melnick & Adelberg, 1981). Prolonged sickness leads to a larger expenditure on health care and an increased risk of death (Tanwar et al., 2014).
Salmonella can be classified into serotypes using a serum agglutination test (Popoff & Le Minor, 1992) and to date more than 2,500 serotypes have been defined (Grimont & Weill, 2007). Monophasic S. Typhimurium variants (1,4,[5],12:i:-) that lack phase two flagellar antigens have become prevalent worldwide and present a significant public health risk through contamination of the food production chain (Gymoese et al., 2017; Petrovska et al., 2016; Yang et al., 2015). Salmonella 1,4,[5],12:i:-isolates represent emergent clones that are often multi-drug resistant (Skarżyńska et al., 2017; Yang et al., 2015) and, along with other monophasic S. Typhimurium, are among the main serotypes found in pig production in northern Thailand (Tadee et al., 2015). Evidence suggests that contaminating strains persist from the farm, through the slaughterhouse, to marketplace (Patchanee et al., 2016). The expansion of the monophasic S. Typhimurium clade (1,4,[5],12:i:-) is associated with the acquisition of the Salmonella genomic island four (SGI-4; up to 87 genes), which contains genes associated with resistance to copper, silver, mercury or arsenic and is thought to have enabled the proliferation of antimicrobial resistance determinants within the pork production industry (Branchu et al., 2019; Patchanee et al., 2016; Tadee et al., 2015). Genomic islands are large, flexible genome elements that carry sets of genes that can confer novel phenotypes, such as heavy metal resistance in this case (Hallstrom & Mccomick, 2015). Furthermore, Salmonella pathogenicity islands (SPIs), a subset of the genomic islands, can also play a pivotal role in virulence and severity (Banaszkiewicz et al., 2019; Elder et al., 2018; Marcus et al., 2000), often associated with specific host niches (Amavisit et al., 2003; Foley et al., 2013). Together, emerging multidrug resistance and enhanced virulence, make successful treatment of salmonellosis patients increasingly difficult (Tanwar et al., 2014).
Global surveillance efforts and development of large genome databases for comparative genomics have enabled quantification of variation within human infection and agricultural isolates (Jolley, Bray & Maiden, 2018; Ribot & Hise, 2016). The improved discriminatory power of next generation sequencing, with increased throughput, scalability and speed has lead to improvements in surveillance and source attribution (Henri et al., 2017; Kwong et al., 2015; Toro et al., 2016). In silico identification and annotation of putative genes, and a better understanding of sequence variation through multilocus sequence typing (MLST) schema have informed epidemiological studies (Bradley et al., 2015; Zankari et al., 2013; Henri et al., 2017; Sheppard, Jolley & Maiden, 2012; Toro et al., 2016).
In this study, we screened isolates from the pork production process in Northern Thailand for the presence of common, high-risk disease-causing lineages. Using gene-by-gene methods, we characterise a collection of nineteen monophasic S. Typhimurium genomes isolated from the pork production chain between 2011–2014 (Patchanee et al., 2016; Tadee et al., 2015). The genetic diversity, presence of antimicrobial resistance, virulence, known plasmid genes and Salmonella pathogenicity islands were explored. We identify infiltration of the globally disseminated, multi-drug resistant ST-34 clone at all stages of pork production and assess the pathogenic potential of each isolate. Our findings will aid surveillance and inform agricultural prevention strategies for salmonellosis in the region.
Materials and Methods
Monophasic S. Typhimurium isolate collection
Monophasic Salmonella Typhimurium were isolated during the period of November 2011 through July 2014 in Chiang Mai and Lamphun provinces, as decribed in the studies of Tadee et al. (2015) and Patchanee et al. (2016). The studies were conducted under ethical approval reference number R18/2554 by the Animal Care and Use Committee committee of Faculty of Veterinary Medicine, Chiang Mai University (FVM-ACUC). Briefly, samples were collected at three stages of pig production: on the farm (n = 10); at the slaughterhouse (n = 6) and at retail from local markets and supermarkets (n = 3) (Table 1).
| Strains ID | Locationa | Source | Sampling Date | ABO-resistance patternb |
|---|---|---|---|---|
| A541006 | Farm 1 | Pig’s faeces | 2 November 2011 | AMP,S,CTX,TE |
| A541007 | Farm 1 | Pig’s faeces | 2 November 2011 | AMP,S,CTX,TE |
| A541011 | Farm 1 | Pig’s faeces | 15 November 2011 | AMP,C,CTX,TE |
| A541012 | Farm 1 | Pig’s faeces | 15 November 2011 | AMP,S,NA,CTX,TE |
| A541013 | Farm 1 | Pig’s faeces | 15 November 2011 | AMP,S,TE |
| A541024 | Farm 2 | Pig’s faeces | 20 December 2011 | AMP,S,CTX,TE |
| A541025 | Farm 2 | Pig’s faeces | 20 December 2011 | AMP,C,S,CTX,TE |
| A543008 | Farm 3 | Stable floor swab | 2 June 2012 | AMP,S,TE |
| A543009 | Farm 1 | Stable floor swab | 27 June 2012 | AMP,C,S,CTX,TE |
| A543010 | Farm 2 | Stable floor swab | 3 July 2012 | AMP,SXT,C,S,CTX,TE |
| 21-JRSP3 | SLH A | Carcass splitter swab | 5 May 2013 | AMP,S,TE |
| 172-BT E22 | SLH B | Carcass swab | 2 June 2013 | AMP,S,TE |
| 173-BT E24 | SLH B | Mesenteric lymphnode | 2 June 2013 | AMP,S,TE |
| 117-LP L4 | SLH C | Lairage area floor swab | 9 June 2013 | AMP,C,S,CTX,TE |
| 60-JRD3 | SLH A | Knife swab | 4 August 2013 | AMP,S,TE |
| 193-BT D11 | SLH B | Carcass swab | 2 September 2013 | AMP,S,TE |
| TP3 | Market X | Butchering pork product | 29 June 2014 | AMP,C,TE |
| RPN | Market Y | Shelf pork product | 29 June 2014 | AMP,S |
| MM2.2 | Market Z | Butchering pork product | 6 June 2014 | AMP,S,TE |
Antimicrobial susceptibility assays
All isolates were tested for their susceptility to antimicrobials using disk diffusion methods following Clinical and Laboratory Standards Institute guidelines (Clinical & Laboratory Standards Institute (CLSI), 2011) by the WHO National Salmonella and Shigella Center, National Institute of Health, Department of Medical Science, Nonthaburi, Thailand. Ten different antimicrobial agents were tested, including ampicillin (AMP) 10 µg, amoxicillin-clavulanic acid (AUG) 20/10 µg, chloramphenicol (C) 30 µg, ciprofloxacin (CIP) 5 µg, cefotaxime (CTX) 30 µg, nalidixic acid (NA) 30 µg, norfloxacin (NOR) 10 µg, streptomycin (S) 10 µg, sulfamethoxazole-trimethoprim (SXT) 23.75/1.25 µg and tetracycline (TE) 30 µg (Table 1).
Whole-genome sequencing
Genomic DNA of 19 monophasic S. Typhimurium isolates was extracted, using a QIAamp DNA mini kit (Qiagen, Crawley, UK) and sequenced on an Illumina MiSeq genome sequencer (Illumina, Cambridge, UK). Nextera XT libraries (Illumina, California, USA) were prepared and short paired-end reads (300 bp) trimmed using TRIMMOMATIC (version 0.33; Bolger, Lohse & Usadel, 2014) and assembled de novo using SPAdes software, with the-careful command (St. Petersburg genome assembler, St. Petersburg, Russia; version 3.10.035) (Bankevich et al., 2012). The average number of contigs was 400 (range: 187–777) for an average total assembled sequence size of 5.02 Mbp (range: 4.87–5.20) with an average genome coverage of ×34 (range 16–46). The average N50 contig length (L50) was 33,091 bp (range: 10,047–84,409) and the average GC content was 52.2% (range: 52.0–52.5). Short read data are archived on the National Center for Biothechnology Information (NCBI) SRA, associated with BioProject accession PRJNA573746 (individual accession numbers in Supplemental File S1). FASTA files of all contiguous assemblies and Supplemental Files are archived at the public data repository figshare (https://doi.org/10.6084/m9.figshare.11305727).
Multilocus sequence typing
Nucleotide sequences for seven housekeeping gene loci, including aroC (chorismate synthase), dnaN (DNA polymerase III subunit beta), hemD (uroporphyrinogenIII cosynthase), hisD (histidinol dehydrogenase), purE (phosphoribosy laminomidazole), sucA (alpha-ketoglutarate dehydrogenase) and thrA (aspartokinase I/homoserine dehydrogenase) were identified. Alleles and subsequent sequence types (ST) were designated using the web-based MLST 2.0 method at the Center for Genomic Epidemiology (https://cge.cbs.dtu.dk/services/MLST/) (Larsen et al., 2012).
Core genome genealogy
An alignment file was constructed from concatenated gene sequences of all core genes (found in ≥95% isolates) from the reference Salmonella Typhimurium genome, SO4698-09 (accession# LN999997.1) using MAFFT (Katoh & Standley, 2013) on a gene-by-gene basis (Méric et al., 2014; Sheppard, Jolley & Maiden, 2012) (size: 4,166,027 bp; Table S1). A gene was considered present in a given genome when its sequence aligned to an SO4698-09 locus with more than 70% sequence identity over at least 50% of sequence length using BLAST. Maximum-likelihood phylogenies were built in IQtree v1.6.8 (Center for Integrative Bioinformatics Vienna, Vienna, Austria) using the GTR + F + I + G4 substitution model. A consensus tree constructed from 1,000 bootstrap trees (Hoang et al., 2018) and visualised on microreact (Argimón et al., 2016).
Whole-genome multilocus sequence typing
Core genome multilocus sequence typing (cgMLST) and pan genome multilocus sequence typing (pgMLST) were performed using BioNumerics version 7.6 (Applied Maths, Ghent, Belgium). A total of 2,978 and 5,022 loci were identified as composing the cgMLST and pgMLST scheme, respectively. Cluster analysis of categorical differences of allelic numbers for cgMLST and pgMLST was applied to generate dendrograms, using the complete linkage algorithms.
Identification of antimicrobial resistance, virulence and plasmid associated genes
Nucleotide sequences of all 19 monophasic S. Typhimurium isolates in FASTA format were compared to antimicrobial resistance genes (ARGs), putative virulence factors and known plasmid genes using ABRICATE 0.9.8 (GNU, Boston, MA, USA), using Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020), the NCBI (Feldgarden et al., 2019), ResFinder 3.0 (Zankari et al., 2012). The virulence factor database (VFDB) (Chen et al., 2016) and PlasmidFinder 2.1 (Carattoli et al., 2014) databases. All databases were accessed at 10 September 2019.
Identification of Salmonella Pathogenicity Islands
The Center for Genomic Epidemiology was also used to identify SPIs in our Salmonella isolates. SPIFinder 1.0 (https://cge.cbs.dtu.dk/services/SPIFinder-1.0/) with default settings of 95% threshold over 60% minimum length were used.
Results
Pig isolates from Thailand are related to a globally disseminated clone
All monophasic S. Typhimurium isolates were sequenced, genomes assembled and typed using an in silico MLST scheme. Each of the 19 isolates were categorised as sequence type 34 (ST-34), with the alleic profile: aroC_10/dnaN_19/hemD_12/hisD_9/purE_5/sucA_9/thrA_2. Isolate genomes were mapped to the S. Typhimurium ST-34 reference, SO4698-09 and homologues of all core genes (Table S2) aligned and used to construct a maximum-likelihood phylogenetic tree (Fig. 1). Isolates were closely related and the sum of all internal branch lengths represented less than 5% of the tree length, indicating that all isolates were closely related to the globally disseminated, livestock-associated ST-34 clone.
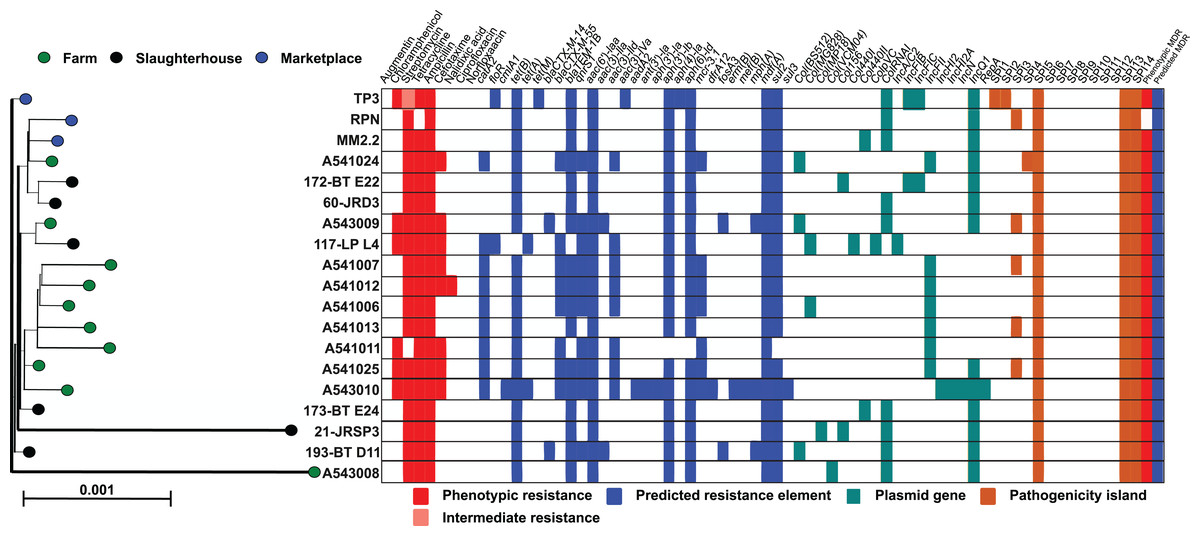
Figure 1: Genetic relatedness of S. enterica serovar Typhimurium isolates.
Genes present in 95% or more of the isolate genomes were aligned (4,166,027 bp) and a maximum-likelihood phylogeny was reconstructed in IQ-TREE with the generalised time reversible substitution model and ultrafast bootstrapping. Scale bar represents genetic distance of 0.001. Each leaf represents a single isolate and is coloured according to sampling location: farm (green), slaughterhouse (black) and retail marketplace (blue). Alongside the tree is a matrix highlighting phenotypic resistance (red), intermediate resistance (light red), presence of predicted AMR genes (blue), plasmid genes (yellow) and presence of Salmonella Pathogenicity Islands (orange).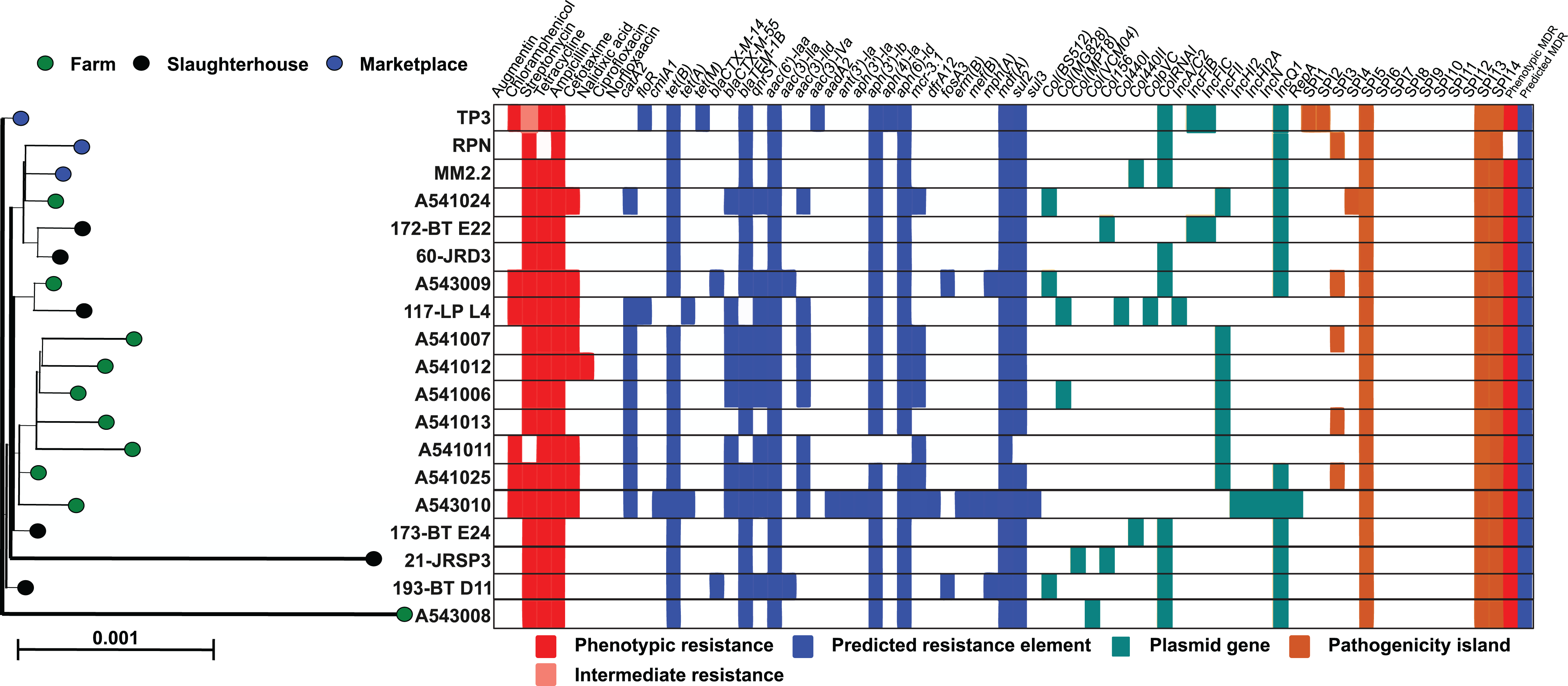{kind=link}
Extended whole genome typing analysis (wgMLST) of the 19 monophasic S. Typimurium genomes was performed for core- (cgMLST; 2,978 genes present in 100% of isolates) and pan- (pgMLST; 5,022 genes) genomes. All isolates were closely related, with a maximum 130 and 170 locus differences between isolates, for cgMLST and pgMLST analyses respectively. Isolates A541011 and A541013 were the most closely related isolates and were collected from the same farm (PD farm), on the same sampling date (15 November 2011) and only differed in 18 core loci. When accessory genes were included in the pan-genome anlaysis, isolates A541006 and A541012 were the most closely related, with 26 loci differences. Visual comparison of the core- and pan-genome MLST dendrograms demonstrates similarity in the clustering of isolates (Fig. S1).
Clonal isolates shared different ARGs
The genome sequences of all 19 monophasic S. Typhimurium isolates were compared using ABRICATE 0.9.8 and known antibiotic resistance genes and alleles from CARD, ResFinder and NCBI databases (Table S3). As there is redundancy in these antibiotic resistance gene sequence databases, we show only results from the ResFinder comparison (Fig. 1). Twenty-five resistance genes were identified, from ten antibiotic classes (Table 2). Eight detected genes (8/25; 32%) were grouped in aminoglycoside classes. All monophasic S. Typhimurium genomes (19/19; 100.00%) harboured aminoglycoside and beta-lactam class resistance genes. Sulphonamide and tetracycline resistance genes were the next most prevelant (18/19; 94.73%), followed by fluoroquinolone (10/19; 52.63%) and phenicol (7/19; 36.84%). Fusidic acid, glycopeptide, nitroimidazole, oxazolidinone and rifampicin associated-resistance genes were not detected in any of the genomes. Putative resistance genes were also detected for colistin (7/19; 36.84%), phosphomycin (2/19; 10.52%) and macrolide-lincosamide-streptogramin B (3/19; 15.78%), although no phenotypic susceptibility tests were performed to support these results.
| Class | Resistance gene | No. of strains carried the gene (%) | No. of strains carried gene grouped in the class (%) |
|---|---|---|---|
| Aminoglycoside | addA1 | 1 (5.26) | 19 (100.00) |
| addA2 | 1 (5.26) | ||
| aac(3)-IIa | 5 (26.32) | ||
| acc(3)-IVa | 1 (5.26) | ||
| acc(3)-IId | 6 (31.58) | ||
| aph(3)-Ib | 18 (94.97) | ||
| aph(4)-Ia | 1 (5.26) | ||
| aph(6)-Id | 18 (94.74) | ||
| Beta-lactam | blaTEM-1B | 17 (89.47) | 19 (100.00) |
| blaCTX-M-14 | 2 (10.53) | ||
| blaCTX-M-55 | 8 (42.11) | ||
| Fluoroquinolone | qnrS1 | 10 (52.63) | 10 (52.63) |
| Phenicol | catA2 | 9 (47.37) | 10 (52.63) |
| cmlA1 | 1 (5.26) | ||
| floR | 2 (10.53) | ||
| Sulphonamide | sul2 | 18 (94.74) | 18 (94.74) |
| sul3 | 1 (5.26) | ||
| Tetracycline | tet(A) | 2 (10.53) | 18 (94.74) |
| tet(B) | 17 (89.47) | ||
| tet(M) | 1 (5.26) | ||
| Trimethoprim | dfrA12 | 1 (5.26) | 1 (5.26) |
| Fosfomycin | fosA4 | 2 (10.53) | 2 (10.53) |
| MLS* | ermB | 1 (5.26) | 3 (15.79) |
| mphA | 3 (15.79) | ||
| Colistin | mcr-3.1 | 7 (36.84) | 7 (36.84) |
Note:
Predicted resistance generally matched the phenotypic susceptibility profiles (Table 3). Nearly half of the antimicrobial classes tested (2/5; beta-lactam and tetracycline) were 100% concordant. In other comparisons, some isolates were predicted to be resistant, but no phenotypic resistance was observed (n = 2 aminoglycoside; n = 9 fluoroquinolone; n = 5 phenicol). Conversely, one isolate was phenicol phenotypically resistant, but no antimicrobial resistance determinant was identified.
Agricultural reservoir isolates contribute to public health risk
Fewer ARG determinants were found in the isolates as pigs passed though the production process (farm: n = 10, mean = 11.8; slaughterhouse n = 6, mean = 8.5; marketplace n = 3, mean = 8.3), although with only a small number of samples strong statistical associations cannot be made. Samples collected from retail pork products were not predicted to confer resistance to macrolides, fosfomycin, fluoroquinolone, diaminopyrimidine or colistin (Fig. 2). Typically, isolates collected from farm animals contained ARGs conferring resistance to a broader range of antimicrobials. Comparison with the Plasmid finder database identified the ColRNAI plasmid (accession: DQ298019) in all marketplace isolates (3/3; 100%), nearly two-thirds (4/6; 66%) of slaughterhouse isolates and only a fifth of farm isolates (2/10; 20%) (Fig. 1). In addition, the IncQ1 plasmid (accession: M28829.1) was identified in multiple samples (13/19; 68%).
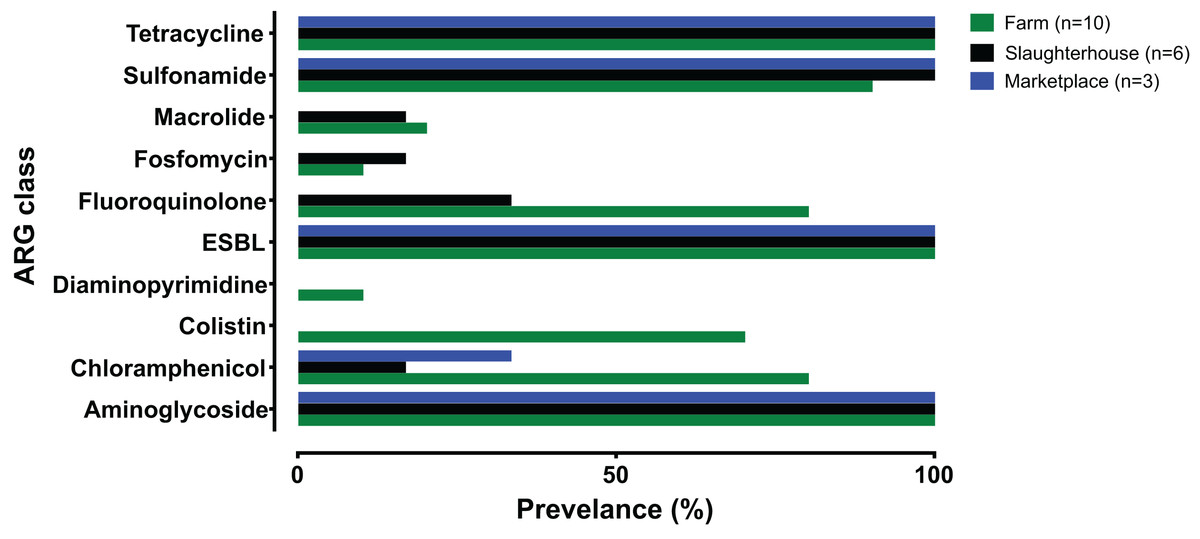
Figure 2: Summary of antimicrobial resistance genes (ARGs) identified, grouped by antibiotic class and collection source.
Farm (green), slaughterhouse (black) and retail marketplace (blue).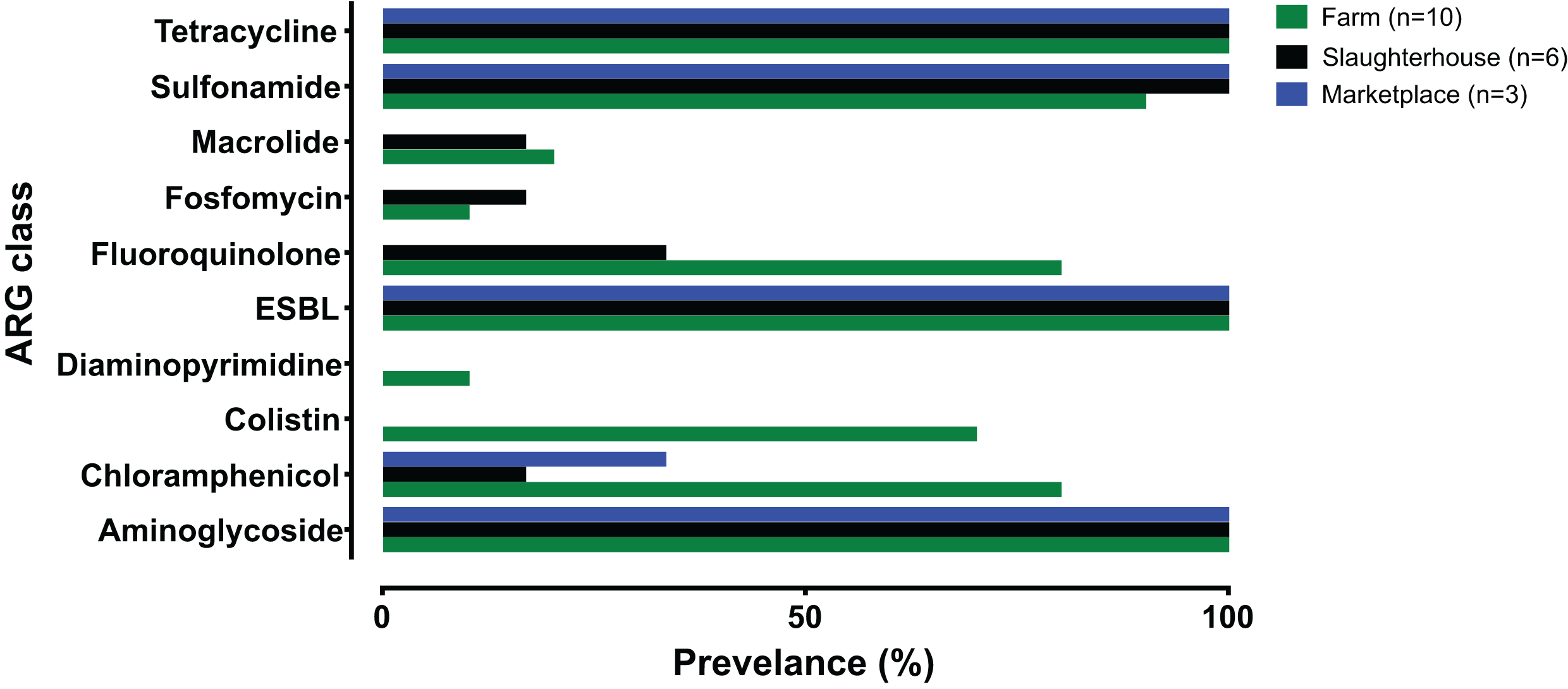{kind=link}
Identification of Salmonella Pathogenicity Island genes
ABRICATE was also used to identify known virulence genes in our monophasic S. Typhimurium isolates with Virulence finder databse (VFDB). In total, 137 different virulence genes were identified, including those from several Salmonella Pathogenicity Islands (SPIs) (Table S3). Seven out of 14 previously described SPIs were detected (Fig. 1A), supported by additional investigation of previously described SPIs using the SPIFinder 1.0 database. Isolates carried three to six SPIs, with more than half (12/19) carrying three islands. The TP3 isolate obtained from a retail pork sample at a local market carried six previously decribed SPIs (SPI-1, SPI-2, SPI-3, SPI-5, SPI-13 and SPI-14) and eleven antibiotic resistance gene determinants from six different antibiotic classes. All the monophasic S. Typhimurium isolates harboured SPI-5, SPI-13 and SPI-14 (100.00%; 19/19 strains), followed by SPI-3, which was carried by nearly one-third of them (31.57%; 6/19 strains).
The large Salmonella genomic island, SGI-4 (up to 87 genes) is often found in ST-34 clones from pigs. Gene-by-gene comparison of our isolates with the Typhimurium reference strain SO4698-09 identified many of the genes linked to carriage of the SGI-4 element (Table S3), including the mercury resistance genes merEDACPTR, which are found near an arsenate resistance operon (aseR, arsD, arsA1, arsB, arsenate reductase, bar N-acetyltransferase, arsA2, arsD2) and silver-copper resistance element (silESRCFBAGP pcoG silE pcoABCDRSE2). Variation has been observed in the SGI-4 genomic island, which has been found to contain up to 87 loci, with 48 described as core. In our collection, at least 84 loci (97%) of the SGI-4 island were identified in all isolates (Table S3).
Discussion
Globalisation and industrialisation of food production has contributed to the emergence of opportunistic pandemic clones (Anderson, 2010). In this study, all of the 19 monophasic S. Typhimurium strains collected from different stages of the pig production process in Northern Thailand were found belong to ST-34, a globally disseminated, multidrug resistant clone closely associated with industrial livestock infections and is often implicated in human food-borne outbreaks (Gymoese et al., 2017; Luo et al., 2019; Zheng & Feng, 2019). Isolates were collected from different stages of the pork production process in Northern Thailand, all of which were found to have been infiltrated by the ST-34 pandemic clone.
We employed a genome-wide, gene-by-gene approach, supported by curated online reference data sets, which provide a standardised MLST nomenclature and the potential to analyse large datasets without specialist bioinformatics expertise or resources (Maiden et al., 2012). Isolates collected from the same place had 18 and 26 loci differences, in core and pan-genome MLST typing schemes, respectively (Fig. S1). Some evidence of heterogeneity was found within the herd, although the sum of all internal branch lengths represented was less than 5% of the core genome maximum-likelihood tree length (Fig. 1), indicating that all isolates were closely related.
Rising AMR in bacteria is a WHO priority, particularly for gastrointestinal pathogens such as Salmonella (World Health Organization (WHO), 2015). The contribution of resistance acquired by livestock associated pathogenic strains on human infection is unclear (Holman & Chenier, 2015; Tanwar et al., 2014; Schwarz, Kehrenberg & Walsh, 2001). Antibiotic use as growth promoters has been banned in European agriculture since 2006, however quinolones and tetracyclines are still available for treatment of livestock (World Health Organization (WHO), 2015). Regulation is less stringent in Thailand, and nearly all the 19 monophasic S. Typhimurium isolates carried resistance elements belonging to aminoglycoside (n = 19), beta-lactam (n = 19), tetracycline (n = 18) and sulphonamide (n = 18) classes which are among the most widely used in industrial pig farming (Van Rennings et al., 2015). Continuous administration at sub-therapeutic level or use as a growth promoter can select for resistance determinants (Holman & Chenier, 2015), and as suggested previously, streptomycin, ampicillin, sulphonamide, tetracycline and similar class antimicrobial are not recommended for clinical salmonellosis treatment (Campioni, Moratto Bergamini & Falcao, 2012; Patchanee et al., 2016; Steeve, John & Dowling, 2013). Several of the isolates collected from farm animals contained colistin resistance elements (7/10, 70%), where colistin may have been used, however these were not found in isolates from later stages of the production process.
Differences were observed between genotype prediction and laboratory phenotype, for example no known antimicrobial resistance determinant could be identified for one isolate that was phenotypically resistant to chloramphenicol (Fig. 1; Table 3). This finding suggests that there may be additional chloramphenicol resistance genes not yet identified or available in the databases. Variation in gene expression levels or synergistic effects among different resistance genes can confound DNA-based predictions and warrants further study (Neuert et al., 2018). Carriage of resistance phenotype and their resistance genes are not routinely tested for some antibiotics (as in our case). This means that isolates carrying genes that can contribute to antibiotic resistance wil be missed (Rosengren, Waldner & Reid-Smith, 2009). Practical and financial constraints can limit the number of selected antimicrobial types that are tested in the laboratory, in silico genotypic identification offers a potential solution to this, and when combined with machine-learning approaches can be hightly accurate (Bradley et al., 2015; Nguyen et al., 2019).
Potential transmission of AMR bacteria between agricultural animals and humans is also a concern, but controversy remains interpreting what constitutes the spread of resistance (Boerlin & Reid-Smith, 2008; Mourkas et al., 2019). Resistance can be transmitted through the food production chain to infect humans, or via recombination and horizontal gene transfer (Yahara et al., 2017). In Salmonella, virulence-associated genes are often mobilised on pathogenicity islands (SPIs) or plasmids and can mediate interactions with the host (Amavisit et al., 2003; Marcus et al., 2000). Only a single isolate in our collection contained SPI-1 and SPI-2, which encode type III protein secretion systems required for intestinal invasion (Lou et al., 2019). The SPI-3 island found in five of our isolates harbours the mgtC gene required for intramacrophage survival (Blanc-Potard et al., 1999). Pathogenicity island-4 (SPI-4), found in one of our isolates, carries a type I secretion apparatus contributing to the colonisation of bovine intestines (Kiss, Morgan & Nagy, 2007). Three SPIs were found in all 19 monophasic S. Typhimurium (SPI-5, SPI-13 and SPI-14). SPI-5 is is often associated with enteropathogenesis, mediating the inflammation and chloride secretion that characterise the enteric phase of disease (Marcus et al., 2000). SPI-13 is required for efficient utilisation of D-glucuronic acid (DGA) and tyramine (TYR) as sole energy sources (Elder et al., 2018). SPI-14 harbours eight open reading frames (ORFs) encoding putative cytoplasmic proteins and is significant during host invasion and colonisation (Jiang et al., 2017).
Conclusions
In this study, we demonstrate that samples tested from multiple stages of the Thai pork production process have been contamined by the globally disseminated, multi-drug resistant ST-34 clone of monophasic S. Typhimurium. Isolates show differences in virulence and antmicrobial resistance profiles, likely affected by acquisition and loss of accessory elements through horizontal gene transfer, mediated by SPIs and plasmids. These clones pose a tangible public health risk, as evidenced by the TP3 strain obtained from a retail market which harbours the SPI-1 pathogenicity island that is important for invasion of host cells and induction of macrophage apoptosis (Amavisit et al., 2003). This emphasises the importance of maintaining hygiene standards and all pork products should be cooked well. Microbial suveillence will help guide intervention measures and provide a reminder that sanitation and hygienic practices at all production levels should be prioritised.
Supplemental Information
Phylogenetic trees of the core genome MLST (cgMLST) compared with pan genome MLST (pgMLST) of 19 Thai monophasic S. Typhimurium.
A set of 2,978 and 5,022 genes was analysed in cgMLST and pgMLST, respectively. Scale distant represents number of loci differences in strains positioned at the common node.