

Abstract
We find stiffness properties of van der Waals heterostructures made of graphene and carbon nanotubes (CNTs) using molecular dynamics (MD) simulations. The variation of the stiffness tensor is studied at different temperatures for structures with changing CNT density, and orientations. Orthotropic structures with unidirectional CNTs have high stiffness ∼300–500 GPa, in one direction. Structures with transverse isotropy show stiffness of ∼150–350 GPa along two mutually perpendicular directions. The orthotropic structures also show greater stiffness with increasing CNT density, while the others are insensitive to it. In general,the stiffness constant values are decrease as temperatures increase. The results can aid in determining useful heterostructure configuration for a particular application and in finding overall stiffness of devices having additional components.
Export citation and abstract BibTeX RIS
1. Introduction
Van der Waals (vdW) heterostructures involving mixed-dimensions (2D + nD, n = 0,1,2,3) have potential applications in electronics, optics, and photovoltaics [1]. The 2D materials generally used are: graphene, hexagonal-boron nitride, transition metal dichalcogenides; 1D materials are single-walled CNTs, organic polymers, inorganic nanowires etc; while 0D materials are nanoparticles like fullerenes and quantum dots.
The focus of this study is 3D heterostructures consisting of graphene and CNTs. These components are known for their extraordinary mechanical, electronic, and thermal properties [2–9]. Several different heterostructure configurations have been proposed and studied, which show enhanced electronic, thermal, and optical properties than the individual components. For example, one paper reports increase in specific capacitance from 190 F g−1 to 318 F g−1 with increasing density of CNTs intercalated between graphene sheets [10]. Another finds that there are gains in efficiency of generation and transport of photocarriers in graphene-CNT films. This improvement in properties occurs because CNTs prevent graphene sheets from restacking, which increases accessible surface area and creates additional transport channels. Graphene-CNT heterostructures, therefore, have great potential for use as batteries [11], catalysts [12], transparent conducting electrodes [13], supercapacitors [14], filtration membranes [15], etc
The existing literature on the mechanical properties of such heterostructures has been focused on complex morphologies like CNT pillars or bridges between graphene sheets [16], Möbius strips of graphene connecting graphene nanoplatelets [17], etc. These morphologies show very good elastic and strength properties because of bonded interactions between graphene and CNTs, however, they are difficult to synthesize. In this paper, we look at the mechanical properties of heterostructures where CNTs are intercalated parallel to graphene, where graphene and CNTs only have weak vdW interactions. These structures are of interest since they approximate heterostructures that have been synthesized experimentally [10, 11, 18]. There has also been a recent computational study exploring synthesis environments that promote CNT intercalation between graphene sheets and self-assembly to generate such vdW heterostructures [19]. Given their relative ease of synthesis and their promising applications as nanodevices, it is worthwhile to quantify their mechanical properties. We use molecular dynamics (MD) simulations to compute the stiffness of these graphene-CNT vdW heterostructures in this work. We study the variation of stiffness at different CNT density and orientation, which is important for synthesis considerations. We also see how the stiffness changes with temperature, which is essential from the device operation standpoint. We compare these values with the theoretical and computed values for high-order pyrolytic graphite (HOPG).
2. Method
For convenience, we refer to the graphene-CNT heterostructures as GNT for the remainder of this paper. Three broad kinds of structures were used in the simulations:
- (i)GNT1 type: this type has all CNTs aligned along the x-axis, parallel to the graphene sheets. Using the notation for composite structures, we say that CNTs have 0/0° arrangement between graphene layers.
- (ii)GNT2 type: this type has bundles of CNTs between alternating graphene sheets aligned along the x- and y-axes, respectively, or have 0/90° arrangement.
- (iii)GNT3 type: this type has -45/45° arrangement of CNTs between graphene sheets.
Some of these structures are shown in figure 1 and dimensions are given in table 1. The subscripts refer to the number of CNTs between two graphene layers, for example, GNT means that there are two CNTs between the lowermost graphene layers and three CNTs between the adjacent graphene layers. Therefore, the structures GNT
means that there are two CNTs between the lowermost graphene layers and three CNTs between the adjacent graphene layers. Therefore, the structures GNT , GNT
, GNT , and GNT
, and GNT , are in increasing order of CNT density. The smallest GNT1 structure was created using a Fortran code in which the atoms in the CNTs and graphene layers were fixed, however, nanotubes were translated and rotated around their axis to find minimum total potential energy configurations. The total potential energy of different configurations was calculated using the Lennard-Jones potential between the CNTs and graphene atoms [20]. The TopoTools package [21] in VMD [22] was used to create larger structures and modify CNT alignment and density. The CNTs are of armchair-type, with a radius of 6.7
, are in increasing order of CNT density. The smallest GNT1 structure was created using a Fortran code in which the atoms in the CNTs and graphene layers were fixed, however, nanotubes were translated and rotated around their axis to find minimum total potential energy configurations. The total potential energy of different configurations was calculated using the Lennard-Jones potential between the CNTs and graphene atoms [20]. The TopoTools package [21] in VMD [22] was used to create larger structures and modify CNT alignment and density. The CNTs are of armchair-type, with a radius of 6.7  . Initial distance between graphene layers is around 19.9
. Initial distance between graphene layers is around 19.9  . Consequently, the graphene-CNT separation is ∼3.4
. Consequently, the graphene-CNT separation is ∼3.4  , at which the vdW interaction energy is the highest [26]. CNTs in the same layer are arranged symmetrically, and the spacing is always greater than 12-13
, at which the vdW interaction energy is the highest [26]. CNTs in the same layer are arranged symmetrically, and the spacing is always greater than 12-13  , which means their initial vdW interaction energy is very low, as found by the study mentioned above.
, which means their initial vdW interaction energy is very low, as found by the study mentioned above.

Figure 1. Heterostructures with different CNT-orientation and density. Top row: GNT1, with 0/0° CNT arrangement; middle row: GNT2, 0/90° CNT arrangement; bottom row: GNT3, with a -45/45° CNT arrangement.The subscripts refer to number of CNTs between alternating layers, starting from the lowest layer.
Download figure:
Standard image High-resolution imageTable 1. Dimensions of lowest CNT density structure of each type used in simulation.
| Type | Dimensions ( ) ) |
|---|---|
GNT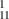 |
9 x 120 x 37 |
GNT |
78 x 119 x 37 |
GNT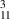 |
78 x 119 x 77 |
Using these heterostructures, we study the change in stiffness constants with:
- (i)varying alignment of CNTs,
- (ii)increasing CNT density (ratio of total CNT atoms to structure volume) and,
- (iii)increasing temperature for a particular alignment and density.
Simulations were carried out using the LAMMPS package [23], with the AIREBO potential [24]. All structures were minimized using the conjugate-gradient method [25] with a force and energy tolerance of 10−7 eV/ and 10−7 eV, respectively. The structures were considered optimized when the total energy per atom converged at a value of 7.4 eV atom−1, which is close to the binding energy of carbon structures. At temperatures higher than zero, i.e. T = 10, 300 K, an NPT simulation was carried out between energy minimization routines. In an NPT simulation, the number of atoms, pressure, and temperature of the system are held constant, while volume and energy are free to change. A simulation timestep of 1 fs and total simulation time of 20 ps was used. Periodic boundary conditions were implemented along the mutually orthogonal in-plane directions. In this step, the non-zero temperature leads to an increase in atomic kinetic energy, causing the CNTs to move around and graphene sheets form ripples around them. As the graphene curves around CNTs, the spacing between graphene layers and CNTs, respectively, decreases at some points. This increases the overall vdW interaction energy of the heterostructures, making them more stable [26]. Additionally, these distortions are desirable as they are consistent with the graphene and graphene-based heterostructures synthesized experimentally, and they have a significant effect on the properties being measured [27, 28].
and 10−7 eV, respectively. The structures were considered optimized when the total energy per atom converged at a value of 7.4 eV atom−1, which is close to the binding energy of carbon structures. At temperatures higher than zero, i.e. T = 10, 300 K, an NPT simulation was carried out between energy minimization routines. In an NPT simulation, the number of atoms, pressure, and temperature of the system are held constant, while volume and energy are free to change. A simulation timestep of 1 fs and total simulation time of 20 ps was used. Periodic boundary conditions were implemented along the mutually orthogonal in-plane directions. In this step, the non-zero temperature leads to an increase in atomic kinetic energy, causing the CNTs to move around and graphene sheets form ripples around them. As the graphene curves around CNTs, the spacing between graphene layers and CNTs, respectively, decreases at some points. This increases the overall vdW interaction energy of the heterostructures, making them more stable [26]. Additionally, these distortions are desirable as they are consistent with the graphene and graphene-based heterostructures synthesized experimentally, and they have a significant effect on the properties being measured [27, 28].
For structures at temperature T = 0 K, stiffness constants were found using the ELASTIC example script in LAMMPS. This script deforms the structure in each direction sequentially by a fixed factor of the structure size. The resulting changes in stress are used to compute the stiffness tensor. For T = 10, 300 K, the minimized structures were used as input for the ELASTIC_T script in LAMMPS, which has a function similar to the ELASTIC script. The script measures the change in the average stress tensor using an NVT simulation when the cell structure undergoes a finite deformation. In an NVT simulation, the number of atoms, volume, and temperature of the system are held constant. The results for all cases were validated by verifying that the stiffness tensor is the same for different values of the deformation parameter.
3. Results
The stiffness tensors are represented using Voigt notation, which has a general form as follows:

The simulations conducted at 0 K help us classify our structures on the basis of symmetry. GNT1 type structures, with unidirectional CNTs, have 9 independent stiffness constants, as shown in table 2a. This implies orthotropic symmetry, or symmetry along three mutually orthogonal planes [32]. The stiffness tensor for these orthotropic materials has the following form:

Table 2. Stiffness constant values (GPa) for different structures and temperatures for symmetry considerations.
| T = 0 K | C11 | C22 | C33 | C12 | C13 | C23 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|
GNT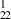 |
378.92 | 299.17 | 246.02 | 76.85 | 82.04 | 89.36 | 24.93 | −49.66 | 111.8 |
GNT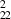 |
325.41 | 350.77 | 89.06 | 59.05 | 53.39 | 44.88 | 24.90 | 28.51 | 128.07 |
GNT |
231.15 | 239.73 | 11.00 | 27.60 | 6.77 | 7.59 | 11.26 | 11.05 | 102.00 |
| (a) | |||||||||
| T = 10 K | C11 | C22 | C33 | C12 | C13 | C23 | C44 | C55 | C66 |
GNT |
239.77 | 54.38 | 2.59 | 9.50 | −1.45 | 6.64 | 1.94 | 7.15 | 71.06 |
GNT |
287.17 | 169.52 | 1.24 | 27.87 | 0.93 | 2.95 | 0.44 | 0.83 | 66.34 |
GNT |
153.07 | 180.15 | 0.63 | 31.54 | 1.71 | 2.67 | 0.78 | 0.69 | 69.21 |
| (b) | |||||||||
| T = 300 K | C11 | C22 | C33 | C12 | C13 | C23 | C44 | C55 | C66 |
GNT |
270.58 | 31.88 | 2.77 | 4.10 | −2.25 | 8.12 | 2.87 | 8.76 | 67.97 |
GNT |
178.03 | 146.84 | 1.08 | 3.29 | 0.31 | 1.46 | 0.14 | 0.94 | 55.02 |
GNT |
92.29 | 240.12 | 1.64 | 12.49 | 3.63 | 4.41 | 2.39 | 1.23 | 74.67 |
| (c) | |||||||||
For GNT2 and GNT3 structures, we see from table 2a that for these structures, C11≈ C22, C44 ≈ C55, and C (C11−C12). Therefore, they have five independent stiffness constants and they have transversely isotropic symmetry, which means they have rotational symmetry about the z-axis in addition to orthotropic symmetry. The stiffness matrix for transversely isotropic materials is as follows [32]:
(C11−C12). Therefore, they have five independent stiffness constants and they have transversely isotropic symmetry, which means they have rotational symmetry about the z-axis in addition to orthotropic symmetry. The stiffness matrix for transversely isotropic materials is as follows [32]:

This is also the form of stiffness tensor for GNT2 and GNT3 type structures, which have perpendicular CNT orientations between alternating graphene layers.
These symmetries are clarified by figures 2(a), (d), and (g). Only two stiffness constants, C11,22, are shown in the graphs, because the rest are either too low, or do not show significant change with temperature or density. We would like to mention that, elastic constants for these structures can be calculated using the material symmetry established here. In the present case, the E1, E2 elastic constant values are very close to C11, C22, and have not been mentioned separately.

Figure 2. Stiffness constants versus CNT density for different structure types at temperature, T = 0, 10, 300 K.
Download figure:
Standard image High-resolution imageThe stiffness computed for HOPG in our simulations is comparable to those reported by ab-initio and experimental studies [33]. From figure 2, we also see that HOPG structures have much higher stiffness than all heterostructures, regardless of CNT density or simulation temperature. Thus, the introduction of CNTs as spacers between graphene sheets leads to lower stiffness compared to HOPG. At temperatures higher than T = 0 K, we find, in general, that stiffness values decrease for all heterostructures and HOPG. This is ascribed to the structural distortions mentioned before, which are shown in figure 3 for GNT after NVT equilibration at temperature T = 300 K. This deterioration of mechanical properties is in agreement with temperature-dependent studies on the mechanical properties of single-layer graphene [29], polycrystalline graphene [30], and monolayer graphene embedded in a polymer matrix [31].
after NVT equilibration at temperature T = 300 K. This deterioration of mechanical properties is in agreement with temperature-dependent studies on the mechanical properties of single-layer graphene [29], polycrystalline graphene [30], and monolayer graphene embedded in a polymer matrix [31].

{kind=link}
{kind=link}
Figure 3. Structural distortions after NVT equilibration at temperature T = 300 K in: (a) GNT , (b) GNT
, (b) GNT , and (c) GNT
, and (c) GNT .
.
Download figure:
Standard image High-resolution image{kind=link}
In GNT1, with unidirectionally oriented CNTs, the C11 values are slightly lower at higher temperatures, while C22 values decrease significantly. There is also a notable increase in C11 values with increasing CNT density, but C22 values do not change. This is due to a trade-off between stiffness reinforcement due to addition of CNTs and stiffness reduction due to structural distortions. For C11 the first factor is much stronger than the latter, whereas C22 is sensitive to the latter. Therefore, in GNT1, we see high stiffness (∼300–500 GPa) only in one direction for T = 10 K, 300 K.
In comparison, for GNT2,3, C11 and C22 have comparable magnitudes at T = 10, 300 K. For GNT2, the 0/90° CNT orientation restricts distortion during minimization as seen in figure 3(b), and its effects are compensated by increase in CNT density for both C11,22. On the other hand, in GNT3, the distortion is not predominant along a particular axis due to the –45/45° CNT orientation, as shown in figure 3(c). Overall, increasing CNT density for GNT2,3 structures does not lead to significant changes in stiffness. However, they have the advantage of having good stiffness values along both mutually orthogonal in-plane directions. GNT2 has slightly higher C11,22 values (∼250-350 GPa) as compared to GNT3 (∼150-200 GPa), because the CNTs in GNT2 are oriented along the 1,2-axis.
These computed stiffness values show that even with the decrease in stiffness compared to HOPG, GNT vdW heterostructures are promising for the various device applications over a range of temperatures. Using these results, an optimized structure can be selected depending on the desired application. Further computational studies can be conducted on the effects of CNT size and chirality on the stiffness of these heterostructures. We can also extend the MD results to find mechanical properties of heterostructures with a dispersed matrix using micromechanics (MM) methods or carry out efficient computations for larger heterostructures using finite element approximations for graphene and CNTs [34].
4. Conclusions
In this study the stiffness constants of graphene-CNT (GNT) heterostructures were computed using MD at different temperatures, CNT densities and orientations. It was found, in general, that stiffness of the heterostructures is much lower than that for graphite, or stacked graphene layers. Orthotropic heterostructures have high stiffness values in the direction of CNT orientation, which increases from about 300 to 500 GPa as CNT density increases, and this is not sensitive to change in temperature. Transversely isotropic heterostructures, with CNTs arranged perpendicular to each other between alternating graphene layers have good stiffness values of 150-350 GPa along two mutually perpendicular axes. Higher stiffness values are seen when CNTs are oriented along loading axes. For transversely isotropic structures, stiffness is not susceptible to change in CNT density, but decreases slightly with increase in temperature. The results of this paper can help select suitable configurations of graphene-CNT heterostructures based on requirements, i.e. if high stiffness is needed along one or more axes, or depending on operating temperature for a particular application, which are important for device design.