
Abstract
Toll-like receptors (TLRs) are a family of pattern recognition receptors that are best-known for their role in host defence from infection. Emerging evidence also suggests that TLRs have an important role in maintaining tissue homeostasis by regulating the inflammatory and tissue repair responses to injury. The development of cancer has been associated with microbial infection, injury, inflammation and tissue repair. Here we discuss how the function of TLRs may relate to these processes in the context of carcinogenesis.
Similar content being viewed by others
Main
The inflammatory response can promote carcinogenesis by multiple mechanisms. These include the anti-apoptotic effect of nuclear factor-κB (NF-κB; a transcription factor commonly engaged in inflammatory conditions), induction of oxidative damage to DNA and the induction of the tissue repair response1,2,3. One of the major challenges in understanding the connection between inflammation and cancer is to identify the triggering events that lead to the inflammatory responses that can promote tumorigenesis.
Inflammation is an adaptive response that is triggered by a variety of abnormal conditions, including infection and tissue injury as well as more subtle alterations of tissue homeostasis. Infection is the best-understood trigger of inflammation, with recognition of microbial pathogens by the host innate immune system initiating a potent inflammatory response4,5. Although this response can be initiated by several types of pattern-recognition receptors (PRRs), the Toll-like receptors (TLRs) are the best-characterized. Here we will discuss TLRs and their role in cancer development.
TLRs recognize microbial ligands
Toll-like receptors are a family of transmembrane receptors that recognize conserved molecular patterns of microbial origin. Accumulating evidence indicates that TLRs also have an important role in tissue repair and tissue injury-induced inflammation. TLR ligands in this case can be either microbial (exogenous) or host-derived (endogenous).
TLRs are best-known for their ability to recognize conserved microbial structures that were originally named PAMPs (pathogen-associated molecular patterns) by Janeway6. Despite their name, PAMPs are common to all microorganisms regardless of their pathogenicity.
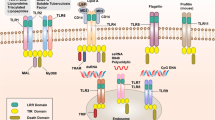
The best-characterized TLR microbial ligands are as follows: lipopolysaccharide (LPS; endotoxin) from Gram-negative bacteria, which stimulates TLR4; bacterial lipoproteins and lipotechoic acid and fungal zymosan, which stimulate TLR1, TLR2 and TLR6; bacterial flagellin, which activates TLR5; a profilin-like molecule from the protozoan Toxoplasma gondii, which activates TLR11; unmethylated CpG motifs present in DNA that function as stimulators of TLR9; double-stranded RNA that activates TLR3; and single-stranded RNA that can stimulate TLR7 and TLR8 (Fig. 1).

TLRs are involved in recognition of microbial and endogenously derived molecular patterns. This occurs both at the plasma membrane and at intracellular compartments. After ligation of TLR ligands either directly or with the help of accessory molecules such as CD14, MD2 (also known as LY96) and CD36, TLRs dimerize and transmit signals throughout the cell by means of adaptor molecules such as myeloid differentiation factor 88 (MYD88) and TRIF. This leads to the activation of multiple cellular phenomena, the best-described of which being the activation signal transduction to the nucleus (such as through activation of nuclear factor-κB (NF-κB), MAPKs and interferon regulatory factors (IRFs). TLR activation leads to regulation of innate and adaptive immune responses, inflammation and tissue repair. LPS, lipopolysaccharide.
In addition to microbial ligands, an increasing number of endogenous ligands are being reported as candidate stimulators of TLRs, in particular of TLR2 and TLR4. These include heat shock proteins (HSP60, HSP70, endoplasmin, HSPB8 and α-crystallin A chain)7,8,9,10,11,12,13, high mobility group box 1 (HMGB1)14,15, uric acid crystals16,17, surfactant protein A18, and various products of the extracellular matrix such as fibronectin19, heparan sulphate20, biglycan21, fibrinogen22, oligosaccharides of hyaluronan23 and hyaluronan breakdown fragments24,25,26.
TLRs control host defence from infection
The most evolutionarily conserved role of TLRs in host defence is the regulation of antimicrobial responses by epithelial cells, the first line of defence at mucosal sites such as the respiratory, gastrointestinal and genitourinary tracts, and the skin. TLRs are involved in the transcriptional and post-transcriptional regulation (proteolytic processing and secretion) of potent antimicrobial factors such as defensins (α and β), phospholipase A2, lysozyme and the regeneration (Reg) family of molecules27,28,29.
TLRs enhance the uptake of microorganisms by phagocytic cells30 and optimize microbial killing through the generation of reactive oxygen and nitrogen intermediates and stimulation of the neutrophil oxidative burst31. Recognition of PAMPs by TLRs leads to the induction of inducible nitric oxide synthetase32 and activation of the NADPH oxidase complex33,34,35, which is important for the production of reactive oxygen and nitrogen intermediates.
TLRs also have a crucial role in mediating leukocyte recruitment to infected tissues. Activation of TLRs on endothelium, either directly or indirectly, leads to the surface expression of E-selectin and intercellular adhesion molecule 1 (ICAM1). These molecules are crucial for leukocyte rolling and adhesion36 and the induction of chemokines that lead to the adhesion of leukocytes to endothelium37.
TLRs are central to the regulation of host protective adaptive immune responses. The stimulation of both T- and B-cell-mediated immune responses by adjuvants containing microbial lysates or products, such as complete Freund's adjuvant, is due mostly to ligation of TLRs38. Activation by TLRs of professional antigen-presenting cells such as dendritic cells is crucial for several processes: T-cell activation39,40,41,42; the processing and presentation of microbial antigens43; upregulation of co-stimulatory molecules such as CD80 and CD86, which are necessary for the activation of naive CD4 T cells44; and the inhibition of regulatory T-cell activity by the production of factors such as interleukin 6 (IL-6)45. TLRs are also crucial for the activation and maturation of the B-cell response during infection and vaccination. Through both T-cell-dependent and T-cell-independent pathways, TLRs regulate the B-cell response by inducing B-cell proliferation, immunoglobulin isotype class switching and somatic hypermutation46. TLRs can also regulate the differentiation and maintenance of T and B cells by the production of IL-12, IL-23 and IL-27. These cytokines induce T-helper type 1 (TH1) and TH17 cell development47, and so help to promote the cell-mediated immune response.
TLR signalling
TLRs localize to several different subcellular compartments and their localization corresponds to the macromolecular nature of the ligands they recognize. Thus, TLRs that recognize lipid and protein ligands are expressed on the plasma membrane (TLR1, TLR2, TLR4, TLR5 and TLR6), whereas TLRs that detect viral nucleic acids are localized to endolysosomal compartments (TLR3, TLR7 and TLR9). TLRs transmit signals through one or more of four adaptor proteins: myeloid differentiation factor 88 (MYD88), TICAM1 (also known as TRIF), TIRAP (also known as MAL), and TICAM2 (also known as TRAM and TIRP). All TLRs (except for TLR3) and IL-1 receptor family members signal through MYD88. TLR3 signals through TICAM1 and TLR4 signals through both the MYD88 and the TICAM1 pathways4.
Stimulation of TLRs leads to activation of NF-κB, MAPKs, JUN N-terminal kinases (JNKs), p38 and ERKs, and interferon regulatory factor (IRF3, IRF5 and IRF7) signalling pathways5. These signals are essential for the classical outcomes of TLR activation: the orchestration of host innate and adaptive immune responses.
TLRs have also been shown to regulate cell death. Activation of the PI3K–Akt signalling pathway by TLRs48,49,50 promotes cell survival, and TLR signalling has also been associated with the increased expression of the anti-apoptotic proteins Bcl-2-related protein A1 (BCL2A1), inhibitor of apoptosis 1 (cIAP1), cIAP2, XIAP and Bcl-2 family members51. However, in the face of obligate intracellular bacteria and viruses, TLRs can induce cell-autonomous apoptosis through TICAM1, FADD, caspase 8 and protein kinase R (PKR)52,53,54,55 or through the induction of type I interferons. In addition, TLRs assist natural killer (NK) cells in the killing of infected cells either by direct stimulation of TLRs on NK cells or the induction of factors such as type I interferons and IL-15 (Ref. 56).
TLRs, tissue repair and regeneration
In addition to their role in mammalian host defence from microbial infection, TLRs are involved in various aspects of tissue homeostasis, including tissue repair and regeneration57,58. Tissue repair involves the restoration of lost cell populations, tissue architecture, blood supply and innervation, and TLRs appear to be involved in this though multiple mechanisms. By providing pro-survival signals and by inhibiting apoptosis, TLRs may dictate the threshold of injury and cell death, thereby limiting the extent of initial tissue injury. In addition, signalling induced after injury, such as the TLR-dependent production of prostaglandins (through cyclooxygenase 2 (COX2) induction) in tissue stroma59,60 or the upregulation of anti-apoptotic factors may provide signals to keep both differentiated and progenitor cells within the tissue alive.
In the nervous system, TLRs regulate neuronal homeostasis by controlling the proliferation and differentiation of adult hippocampal neurons in the brain61. In this context, TLRs have also been shown to inhibit neurite outgrowth62. The repair of axons, spinal cord or brain tissue that has been damaged as a result of crush injury is also coordinated by TLRs63,64,65. In the large intestine, even in the absence of overt infection or injury, TLR activation mediates proper epithelial barrier function by strengthening tight junctions and inducing cytoprotective factors66,67,68. TLR signalling also mediates the repair of damaged tissue secondary to multiple types of colonic injury, including damage as a result of infection or exposure to chemicals or radiation66,69,70,71,72. This leads to the repositioning of tissue-resident prostaglandin-secreting stromal cells, which then trigger colonic epithelial progenitors to proliferate at crypt bases60. In the lung, basal inhibition of apoptosis through signalling of TLR2 and TLR4 maintains epithelial homeostasis24 and also protects from bleomycin- and hyperoxia-induced lung injury24,73. TLR signalling is also necessary for liver regeneration after partial hepatectomy74,75 and full thickness wound healing in the skin76. TLRs have been shown to regulate the compensatory proliferation of parenchymal cells after injury in the liver, colon and central nervous system66,70,74,75,77.
What ligands activate TLRs to induce tissue repair and to maintain tissue homeostasis? At sites colonized by the indigenous microflora, such as the colon, microbe-derived ligands stimulate TLR-dependent tissue homeostasis during the steady state and following injury66,70. Ligands derived from the indigenous flora may also be important in the liver owing to the presence of microbial ligands for TLRs in entero-hepatic circulation78. In sterile sites, TLRs are likely to be activated by endogenous ligands that are derived from necrotic cells, such as HMGB1 (Refs 79,80) or extracellular matrix components that are generated as a result of tissue injury81.
TLRs and cancer
TLRs as negative regulators of cancer. In the 18th century Deidier observed a positive correlation between infection and the remission of malignant disease, making the first inference that microbes could have anticancer properties82. At the end of the 19th century, William Coley observed that repeated injections of a mixture of bacterial toxins from the Gram-positive bacterium Streptococcus pneumoniae and the Gram-negative bacterium Serratia marcescens served as efficient anti-tumour therapeutic agents, providing evidence that microbial products, rather than infection itself, may mediate an anti-tumour effect83. It was later discovered by Shear and Turner that LPS was the “haemorrhage producing fraction” of Coley's toxin that accounted for its anti-tumour effect82. As LPS stimulates TLR4, these results suggest that the anti-tumour effect of Coley's toxin was a result of TLR activation.
Other microbe-derived therapeutics with anti-tumour activity can be linked to their ability to activate TLR. OK-432, a lyophilized preparation of group A streptococcus84 that is used in the treatment of cervical, gastric and oral squamous cell carcinoma85,86,87 was recently shown to stimulate TLR4 (Refs 88,89). Mycobacterium bovis bacillus Calmette–Guérin (BCG; a potent activator of TLR2- and TLR4-dependent signalling)90,91 has been used for 30 years as an effective treatment of bladder cancer through the intravesicular injection of these mycobacteria92.
Potent anticancer effects against established tumours in both mice and humans after the administration of purified ligands for TLRs have been demonstrated82,93 as a result of local (at the site of the tumour) and systemic delivery93. Administration of LPS has been used in Phase II clinical trials for the treatment of colorectal and lung cancer94 and leads to tumour regression when directly injected into adoptively transferred tumours95. This effect has also been shown on injection of flagellin96. Locally, application of synthetic ligands for TLR7 and TLR8, such as imiquimod, are under investigation for the treatment of skin cancer and chronic lymphocytic leukaemia97,98,99 as is the TLR9 ligand, CpG, for the treatment of brain, skin and renal cancer and lymphoma93,100.
TLR agonists might mediate their anti-tumour activity through a multitude of mechanisms. High doses of TLR agonists, especially those such as poly(IC) that stimulate TLR3, can lead to apoptosis and have been shown to directly kill both tumour cells and ancillary cells of the tumour microenvironment, such as the vascular endothelium101,102,103,104. TLR activation may also cause tumours to regress by increasing vascular permeability82 and by directly or indirectly recruiting leukocytes, resulting in tumour lysis by NK and cytotoxic T-cells.
One of the most appreciated functions of TLRs in cancer therapy is stimulation of the adaptive immune system. In these studies, tolerance to tumour self-antigens is broken, presumably by TLR-mediated upregulation of co-stimulatory signals to the adaptive immune response, a property known as adjuvanticity. This has been used in cancer vaccines, as targets of gene therapy and in raising anti-tumour antigen-specific T cells in vitro for adoptive transfer93.
Does activation of TLRs have a physiological (not only an iatrogenic) role in inhibiting tumorigenesis? In the majority of studies, exogenous TLR agonists have been used to induce anticancer T-cell responses. The responses are often not achieved under physiological circumstances. However, two recent studies have suggested a more physiological role of TLRs in inducing anti-tumour T-cell responses79. In one study, the ability of numerous chemotherapeutic agents to kill established, adoptively transferred tumours was decreased in TLR4- and MYD88-deficient mice79. The authors demonstrate that HMGB1 binds to TLR4 and that HMGB1, which is released by chemotherapy-induced cell death, can activate TLR4 and induce anti-tumour T-cell immunity79. In a second report, C3H/HeJ mice with a loss-of-function mutation in TLR4 that were treated with DMBA to induce skin tumours developed more tumours than wild-type mice, perhaps owing to decreased activation of interferon-γ-dependent anti-tumour T-cell responses105.
The major physiological role that TLRs have in cancer may be preventing infection by microbial pathogens that are associated with its development. TLRs are important in the recognition of microbial pathogens such as Epstein–Barr virus106, hepatitis B and C viruses107,108,109,110, human papilloma virus111 and Helicobacter pylori112,113, all of which are important aetiological agents of human cancer.
Classically, the ability of TLR signalling to activate the adaptive immune system has led to attempts to harness this response against cancer cells through the use exogenous administration of TLR ligands. More research is needed to determine the role of microbial and endogenous TLR ligands in inhibiting tumorigenesis in both infectious and non-infectious situations.
TLR stimulation drives tumorigenesis. The original idea that stimulation of TLRs has a positive role in tumorigenesis came from reports demonstrating that TLR ligands augment the growth of adoptively transferred tumours96,114,115,116,117. For example, systemic LPS administration increased migration, invasion and angiogenesis at secondary sites of an intravenously injected metastatic mammary adenocarcinoma cell line115. Similarly, increased rates of proliferation and decreased rates of apoptosis were evident in metastatic tumours that were formed after the adoptive transfer of a colonic adenocarcinoma cell line followed 9 days later by an intraperitoneal injection of LPS116. Indeed, TLR stimulation in a variety of tumour cell lines leads to increased survival and proliferation in vitro. Isolated plasma cells from patients with multiple myeloma were shown to express an increased repertoire of TLR compared with plasma cells from healthy donors118,119. Stimulation of these cells with various TLR ligands led to increased proliferation in part owing to autocrine secretion of IL-6 (Refs 118,119). By decreasing endogenous expression of TLRs in tumour cell lines it was demonstrated that TLR4 was required for the optimal growth of adoptively transferred tumour cells lines (even in the absence of exogenous LPS administration) and that intratumoral administration of Listeria monocytogenes induced TLR2 signalling in tumour cells and promoted their growth117,120.
TLR ligand administration might also act on host cells to enhance the growth of adoptively transferred tumour cells. In a model in which systemic administration of LPS enhanced the growth of adoptively transferred cells, it was shown that TLR4 signalling in the recipient was required for LPS-induced tumour growth. The suggested mechanism involved a host-dependent increase in circulating levels of TNF that led to the upregulation of NF-κB-regulated anti-apoptotic factors, such as BCL-XL, cIAP1 and cIAP2, in the tumour cells116.
The studies cited above have demonstrated a role of TLRs in promoting tumour survival and growth using adoptive transfer methods. Recently, however, data have indicated that TLRs (and IL-1–IL-18R signalling) have a crucial role in the development of tumours as they arise in their natural microenvironment, thus revealing a previously unknown aspect of tumorigenesis. Mice injected with the mutagen diethylnitrosamine develop liver carcinomas that are dependent on both mutagen-induced cell death and compensatory proliferation. It has been suggested that the response of stromal cells such as tissue-resident macrophages to the death of hepatocytes is crucial to the proliferation and expansion of pre-cancerous cells and tumour promotion121. This promotion is the result of the NF-κB-dependent production of inflammatory mediators such as IL-6 following recognition of necrotic hepatocytes by tumour stroma121,122. Recently, it was shown that MYD88 is crucial for the promotion of diethylnitrosamine-induced hepatocellular tumours122. In response to hepatocyte cell death, MYD88 signalling was responsible for the activation of NF-κB and for the production of factors such as IL-6 (Ref. 122).
MYD88 has also recently been shown to be crucial for tumour promotion in models of spontaneous (ApcMin/+) and carcinogen-induced (azoxymethane) intestinal tumorigenesis123. ApcMin/+ mice are heterozygous for a mutant allele of the tumour suppressor adenomatous polyposis coli (APC)124. After loss of heterozygosity (LOH) at the APC locus, small foci of initiated intestinal epithelial cells form macroscopic tumours. This process is dependent on factors derived from epithelial cells, such as matrix metalloproteinase 7 (MMP7), and the tumour stroma, such as COX2 (Refs 125,126,127). ApcMin/+ mice that are deficient in MYD88 have both a decreased incidence and size of tumours compared with ApcMin/+ wild-type mice123. The effect of MyD88-dependent signalling was independent of initiation as the frequency of microademomas (a proxy for LOH) was similar between MYD88-deficient and MYD88-sufficent ApcMin/+ mice within a litter. Insight into the mechanism by which MYD88 regulated tumorigenesis came from analysis of positive regulators of tumorigenesis, which demonstrated that MYD88 regulated the expression of COX2, MMP7 and cytosolic phospholipase A2 (Ref. 123), which are important in many aspects of tumour growth125,126,127,128.
A recent report demonstrated that, in addition to its action in the liver and intestinal tract, MYD88 is a crucial positive regulator of chemically induced tumours of both skin and connective tissue129. MYD88-deficient mice formed fewer tumours upon administration of DMBA and TPA or 3′-methylcholanthrene (MCA), which led to the development of skin papillomas and sarcomas, respectively129.
These recent studies indicate that TLR signalling contributes to the growth of tumours in numerous organs and thus may represent a general principle of tumorigenesis. As MYD88 is also activated by IL-1–IL-18R activity, it is possible that this pathway also contributes to tumorigenesis in these models.
Whether TLRs are involved in tumour initiation is not yet clear. The data from the ApcMin/+ mice indicate that they are not. However, the Il10−/− mouse model of colitis-associated carcinoma shows that colitis in this context is dependent on TLR recognition of the intestinal microflora. Thus, as tumour initiation is secondary to colitis, a role for TLRs seems likely130. A formal role of TLRs in initiation with concatenate inflammation is yet to be determined; however, one can envision several possible roles for TLRs in initiation. Chronic and unregulated stimulation of TLRs could lead to damage and mutation of DNA and aberrant chromosomal translocation by inducing free radicals or activation-induced deaminase, respectively112,113,131,132. Furthermore, induction of factors such as macrophage migration inhibiting factor (MIF) and BCL-6 by TLRs may impair the DNA damage response through inhibition of p53-mediated growth arrest and apoptosis133,134, thereby driving tumour initiation.
Importantly, the experiments discussed above indicate a link between tissue repair and tumorigenesis at the level of molecular recognition of tissue injury. It remains to be determined whether the TLR-mediated homeostatic response to tissue injury that is associated with tumorigenesis orchestrates processes such as angiogenesis that are ancillary to tumour promotion. TLR activation is known to stimulate angiogenesis in vitro135 through the expression of pro-angiogenic factors such as IL-8, vascular endothelial growth factor and MMPs, and there is in vivo evidence that TLRs might regulate the angiogenic switch76. Futhermore, TLR signalling in tumour cells, the vascular endothelium or other cells may aid various stages of progression and metastasis. In addition to regulating angiogenesis, TLR signalling has been shown to augment tumour cell adhesion and invasion and increase vascular permeability136, although a role for TLRs in the natural events of metastasis has yet to be determined.
PRRs and tumorigenesis
Over the past decade we have learned that there are a limited number of conserved modules by which metazoans recognize and orchestrate a response to different classes of microbes137. It is likely that there will also be a limited number of modules by which organisms respond to and repair different types of injured tissues. For example, we know that there are hypoxia-dependent and hypoxia-independent homeostatic pathways of tumour-associated neoangiogenesis138. Are TLRs a special family of PRRs that are involved in homeostatic aspects of tumorigenesis because they have endogenous ligands, have an ancient role in development and are crucial to adult mammalian tissue repair? Or will other families of PRRs involved in microbial and tissue injury recognition systems act similarly to TLRs during certain types of tumorigenesis? The host response to parasites is often associated with tissue remodelling and results in certain classes of inflammatory responses such as the recruitment of mast cells137, which can be important for tumour growth and survival139,140. Could a common pattern recognition module couple mast cell recruitment in these physiological and pathophysiological host responses? Are there type I and type II classes of tumour-associated inflammation driven by instructions from different types of pattern recognition molecules? If pattern recognition responses are required for tumorigenesis and these are homeostatic responses, then perhaps we can learn how many of these types of responses there are for a given tissue or tumour and therefore determine how robust these processes are to inhibition141. If there are a restricted number of ways for a tissue to induce the inflammatory and repair response, than perhaps there are a limited number of ways for a tumour to develop.
Concluding remarks
It has been known for some time now that tumorigenesis (especially in the case of solid tumours) bears the stamp of inflammation and tissue repair. Tumour promotion and progression are associated with the production of numerous inflammatory mediators, including cytokines and chemokines, the recruitment and activation of leukocytes, particularly mast cells, macrophages and neutrophils, and the orchestration of ancillary processes such as neoangiogenesis and tissue remodelling.
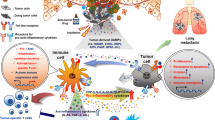
Why is the inflammatory response so tightly coupled with tumorigenesis? As noted above, there are likely to be several unrelated reasons that underlie this connection. In this regard, it is important to distinguish between inflammation-induced cancers and cancer-induced inflammation. Thus, infection- or injury-induced inflammation can promote tumorigenesis owing to chronic tissue damage with subsequent induction of tissue repair. On the other hand, tumour growth can mimic tissue damage (in the case of solid tumours) and this can trigger TLR-dependent inflammatory responses (Fig. 2). Likewise, tumour growth — even without overt tissue damage — is sensed (in some cases by TLRs) as an abnormal occurrence and thus triggers inflammation by mechanisms that are still poorly defined.

In organs such as the lung, brain and colon, activation of TLR signalling at the steady state maintains tissue architecture (a). In the face of tissue injury due either to infection or aseptic noxious stimuli, signalling of TLRs is instrumental in inducing tissue repair. This signalling can be caused by the colonizing microflora, infectious microbes or endogenous ligands such as extracellular matrix or intracellularly derived molecules. The eradication of the noxious stimulus and/or repair of the tissue negatively feeds back on this circuit to suppress the tissue repair programme (b). However in the presence of deregulated infection, inflammation and/or tissue injury as occurs during various stages of tumorigenesis, the unregulated TLR-regulated tissue repair response can drive tumour growth and progression in a positive feedback of unregulated tissue injury and repair (c).
As sensors of cell death and tissue remodelling, TLRs may have a universal role in cancer. Cell death, tissue injury and remodelling may be unavoidable consequences of tumour development and are also associated with a plethora of environmental risk factors for cancer, such as the effects of chemicals and physical trauma. Thus, cancer-induced inflammation is not a stochastically acquired and selected property. Rather, cancer-induced inflammation may be a normal response of the host to the tissue injury and malfunction that is caused by tumour growth. This cancer-associated tissue injury does not resolve itself but may actually be perpetuated by the homeostatic inflammatory and tissue repair response142 (Fig. 2).
Our understanding of the role of TLRs (and other PRRs) in cancer is primitive. Yet, it is clear from the results of animal studies that TLRs have an important part in cancer development. This conjecture is supported by the association of numerous polymorphisms in TLRs with human cancer in a variety of organs including the nasopharynx, stomach, prostate, breast, blood and colon143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159. Future studies will be necessary to delineate the mechanisms by which gain- or loss-of-function polymorphisms in TLRs correlate with risk or protection from cancer.
Harnessing TLRs for cancer immunotherapy and vaccines is promising. However, in a variety of circumstances the activation of TLRs may aid the development of tumours, so targeting the TLRs will not be a straightforward process. Further understanding the role of TLRs and other PRRs in tumorigenesis should provide interesting insights into cancer development. A major challenge for the future will be to dissect the key parameters that determine the outcome of TLR stimulation on tumour initiation and progression.
References
Balkwill, F. & Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 357, 539–545 (2001).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
Karin, M. & Greten, F. R. NF-κB: linking inflammation and immunity to cancer development and progression. Nature Rev. Immunol. 5, 749–759 (2005).
Takeda, K., Kaisho, T. & Akira, S. Toll-like receptors. Annu. Rev. Immunol. 21, 335–376 (2003).
Lee, M. S. & Kim, Y. J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76, 447–480 (2007).
Janeway, C. A. Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54 (Pt 1), 1–13 (1989).
Ohashi, K., Burkart, V., Flohe, S. & Kolb, H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164, 558–561 (2000).
Vabulas, R. M. et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the Toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 276, 31332–31339 (2001).
Vabulas, R. M. et al. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 277, 15107–15112 (2002).
Asea, A. et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 277, 15028–15034 (2002).
Dybdahl, B. et al. Inflammatory response after open heart surgery: release of heat-shock protein 70 and signaling through Toll-like receptor-4. Circulation 105, 685–690 (2002).
Vabulas, R. M. et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J. Biol. Chem. 277, 20847–20853 (2002).
Roelofs, M. F. et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J. Immunol. 176, 7021–7027 (2006).
Park, J. S. et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 290, C917–924 (2006).
Park, J. S. et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377 (2004).
Liu-Bryan, R., Scott, P., Sydlaske, A., Rose, D. M. & Terkeltaub, R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 52, 2936–2946 (2005).
Liu-Bryan, R., Pritzker, K., Firestein, G. S. & Terkeltaub, R. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J. Immunol. 174, 5016–5023 (2005).
Guillot, L. et al. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J. Immunol. 168, 5989–5992 (2002).
Okamura, Y. et al. The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276, 10229–10233 (2001).
Johnson, G. B., Brunn, G. J., Kodaira, Y. & Platt, J. L. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J. Immunol. 168, 5233–5239 (2002).
Schaefer, L. et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Invest. 115, 2223–2233 (2005).
Smiley, S. T., King, J. A. & Hancock, W. W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167, 2887–2894 (2001).
Termeer, C. et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 195, 99–111 (2002).
Jiang, D. et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nature Med. 11, 1173–1179 (2005).
Taylor, K. R. et al. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 279, 17079–17084 (2004).
Taylor, K. R. et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 282, 18265–18275 (2007).
Mukherjee, S., Vaishnava, S. & Hooper, L. V. Multi-layered regulation of intestinal antimicrobial defense. Cell. Mol. Life Sci. 65, 3019–3027 (2008).
Ganz, T. Defensins: antimicrobial peptides of innate immunity. Nature Rev. Immunol. 3, 710–720 (2003).
Brandl, K., Plitas, G., Schnabl, B., DeMatteo, R. P. & Pamer, E. G. MyD88-mediated signals induce the bactericidal lectin RegIII γ and protect mice against intestinal Listeria monocytogenes infection. J. Exp. Med. 204, 1891–1900 (2007).
Blander, J. M. & Medzhitov, R. Regulation of phagosome maturation by signals from toll-like receptors. Science 304, 1014–1018 (2004).
Nathan, C. & Shiloh, M. U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl Acad. Sci. USA 97, 8841–8848 (2000).
Kleinert, H., Pautz, A., Linker, K. & Schwarz, P. M. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol. 500, 255–266 (2004).
Park, H. S. et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J. Immunol. 173, 3589–3593 (2004).
Laroux, F. S., Romero, X., Wetzler, L., Engel, P. & Terhorst, C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of Gram-negative bacteria. J. Immunol. 175, 5596–5600 (2005).
Remer, K. A., Brcic, M. & Jungi, T. W. Toll-like receptor-4 is involved in eliciting an LPS-induced oxidative burst in neutrophils. Immunol. Lett. 85, 75–80 (2003).
Picker, L. J. & Butcher, E. C. Physiological and molecular mechanisms of lymphocyte homing. Annu. Rev. Immunol. 10, 561–591 (1992).
Laudanna, C., Kim, J. Y., Constantin, G. & Butcher, E. Rapid leukocyte integrin activation by chemokines. Immunol. Rev. 186, 37–46 (2002).
Schnare, M. et al. Toll-like receptors control activation of adaptive immune responses. Nature Immunol. 2, 947–950 (2001).
Sallusto, F. et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 28, 2760–2769 (1998).
Forster, R. et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99, 23–33 (1999).
Dieu, M. C. et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 188, 373–386 (1998).
Gunn, M. D. et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 189, 451–460 (1999).
Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nature Immunol. 5, 987–995 (2004).
Medzhitov, R., Preston-Hurlburt, P. & Janeway, C. A. Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 (1997).
Pasare, C. & Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 (2003).
Gerondakis, S., Grumont, R. J. & Banerjee, A. Regulating B-cell activation and survival in response to TLR signals. Immunol. Cell Biol. 85, 471–475 (2007).
Reiner, S. L., Sallusto, F. & Lanzavecchia, A. Division of labor with a workforce of one: challenges in specifying effector and memory T cell fate. Science 317, 622–625 (2007).
Martin, M., Katz, J., Vogel, S. N. & Michalek, S. M. Differential induction of endotoxin tolerance by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli. J. Immunol. 167, 5278–5285 (2001).
Martin, M., Rehani, K., Jope, R. S. & Michalek, S. M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nature Immunol. 6, 777–784 (2005).
Monick, M. M. et al. Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of β-catenin. J. Immunol. 166, 4713–4720 (2001).
Salaun, B., Romero, P. & Lebecque, S. Toll-like receptors' two-edged sword: when immunity meets apoptosis. Eur. J. Immunol. 37, 3311–3318 (2007).
Aliprantis, A. O., Yang, R. B., Weiss, D. S., Godowski, P. & Zychlinsky, A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 19, 3325–3336 (2000).
Ruckdeschel, K. et al. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-β, but not MyD88, in bacteria-infected murine macrophages. J. Immunol. 173, 3320–3328 (2004).
Hsu, L. C. et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428, 341–345 (2004).
De Trez, C. et al. TLR4 and Toll-IL-1 receptor domain-containing adapter-inducing IFN-β, but not MyD88, regulate Escherichia coli-induced dendritic cell maturation and apoptosis in vivo. J. Immunol. 175, 839–846 (2005).
Smyth, M. J., Dunn, G. P. & Schreiber, R. D. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 90, 1–50 (2006).
Larsen, P. H., Holm, T. H. & Owens, T. Toll-like receptors in brain development and homeostasis. Sci. STKE 2007, pe47 (2007).
Michelsen, K. S. & Arditi, M. Toll-like receptors and innate immunity in gut homeostasis and pathology. Curr. Opin. Hematol. 14, 48–54 (2007).
Fukata, M. et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology 131, 862–877 (2006).
Brown, S. L. et al. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J. Clin. Invest. 117, 258–269 (2007).
Rolls, A. et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nature Cell Biol. 9, 1081–1088 (2007).
Ma, Y. et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J. Cell Biol. 175, 209–215 (2006).
Kigerl, K. A. et al. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J. Neurochem. 102, 37–50 (2007).
Babcock, A. A. et al. Toll-like receptor 2 signaling in response to brain injury: an innate bridge to neuroinflammation. J. Neurosci. 26, 12826–12837 (2006).
Kim, D. et al. A critical role of Toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem. 282, 14975–14983 (2007).
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Cario, E., Gerken, G. & Podolsky, D. K. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 127, 224–238 (2004).
Cario, E., Gerken, G. & Podolsky, D. K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 132, 1359–1374 (2007).
Fukata, M. et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G1055–G1065 (2005).
Pull, S. L., Doherty, J. M., Mills, J. C., Gordon, J. I. & Stappenbeck, T. S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl Acad. Sci. USA 102, 99–104 (2005).
Araki, A. et al. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J. Gastroenterol. 40, 16–23 (2005).
Gibson, D. L. et al. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell. Microbiol (2007).
Zhang, X. et al. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J. Immunol. 175, 4834–4838 (2005).
Seki, E. et al. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology 41, 443–450 (2005).
Campbell, J. S. et al. Proinflammatory cytokine production in liver regeneration is Myd88-dependent, but independent of Cd14, Tlr2, and Tlr4. J. Immunol. 176, 2522–2528 (2006).
Macedo, L. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am. J. Pathol. 171, 1774–1788 (2007).
Zhang, Z. & Schluesener, H. J. Mammalian Toll-like receptors: from endogenous ligands to tissue regeneration. Cell. Mol. Life Sci. 63, 2901–2907 (2006).
Seki, E. et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nature Med. 13, 1324–1332 (2007).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature Med. 13, 1050–1059 (2007).
Scaffidi, P., Misteli, T. & Bianchi, M. E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 (2002).
Jiang, D., Liang, J., Li, Y. & Noble, P. W. The role of Toll-like receptors in non-infectious lung injury. Cell Res. 16, 693–701 (2006).
Garay, R. P. et al. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur. J. Pharmacol. 563, 1–17 (2007).
Coley, W. B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin. Orthop. Relat. Res., 3–11 (1991).
Okamoto, H., Shoin, S., Koshimura, S. & Shimizu, R. Studies on the anticancer and streptolysin S-forming abilities of hemolytic streptococci. Jpn. J. Microbiol. 11, 323–326 (1967).
Kikkawa, F. et al. Randomised study of immunotherapy with OK-432 in uterine cervical carcinoma. Eur. J. Cancer 29A, 1542–1546 (1993).
Maehara, Y. et al. Postoperative immunochemotherapy including streptococcal lysate OK-432 is effective for patients with gastric cancer and serosal invasion. Am. J. Surg. 168, 36–40 (1994).
Sato, M. et al. Therapy for oral squamous cell carcinoma by tegafur and streptococcal agent OK-432 in combination with radiotherapy: association of the therapeutic effect with differentiation and apoptosis in the cancer cells. Apoptosis 2, 227–238 (1997).
Okamoto, M. et al. Mechanism of anticancer host response induced by OK-432, a streptococcal preparation, mediated by phagocytosis and Toll-like receptor 4 signaling. J. Immunother. (1997). 29, 78–86 (2006).
Hironaka, K., Yamaguchi, Y., Okita, R., Okawaki, M. & Nagamine, I. Essential requirement of toll-like receptor 4 expression on CD11c+ cells for locoregional immunotherapy of malignant ascites using a streptococcal preparation OK-432. Anticancer Res. 26, 3701–3707 (2006).
Tsuji, S. et al. Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette-Guerin: involvement of toll-like receptors. Infect. Immun. 68, 6883–6890 (2000).
Uehori, J. et al. Dendritic cell maturation induced by muramyl dipeptide (MDP) derivatives: monoacylated MDP confers TLR2/TLR4 activation. J. Immunol. 174, 7096–7103 (2005).
Razack, A. H. Bacillus Calmette-Guerin and bladder cancer. Asian J. Surg. 30, 302–309 (2007).
Krieg, A. M. Development of TLR9 agonists for cancer therapy. J. Clin. Invest. 117, 1184–1194 (2007).
Otto, F. et al. Phase II trial of intravenous endotoxin in patients with colorectal and non-small cell lung cancer. Eur. J. Cancer 32A, 1712–1718 (1996).
Chicoine, M. R. et al. The in vivo antitumoral effects of lipopolysaccharide against glioblastoma multiforme are mediated in part by Toll-like receptor 4. Neurosurgery 60, 372–380; discussion 381 (2007).
Sfondrini, L. et al. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J. Immunol. 176, 6624–6630 (2006).
Scheel, B. et al. Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur. J. Immunol. 36, 2807–2816 (2006).
Stockfleth, E. et al. The use of Toll-like receptor-7 agonist in the treatment of basal cell carcinoma: an overview. Br. J. Dermatol. 149 (Suppl. 66), 53–56 (2003).
Spaner, D. E. & Masellis, A. Toll-like receptor agonists in the treatment of chronic lymphocytic leukemia. Leukemia 21, 53–60 (2007).
Carpentier, A. et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 8, 60–66 (2006).
Salaun, B., Coste, I., Rissoan, M. C., Lebecque, S. J. & Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 176, 4894–4901 (2006).
El Andaloussi, A., Sonabend, A. M., Han, Y. & Lesniak, M. S. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia 54, 526–535 (2006).
Haimovitz-Friedman, A. et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J. Exp. Med. 186, 1831–1841 (1997).
Nogueras, S. et al. Coupling of endothelial injury and repair. An analysis using an in vivo experimental model. Am. J. Physiol. Heart Circ. Physiol. 294, H708–H713 (2008).
Yusuf, N. Protective role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 68, 615–622 (2008).
Gaudreault, E., Fiola, S., Olivier, M. & Gosselin, J. Epstein–Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 81, 8016–8024 (2007).
Broering, R. et al. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J. Hepatol 48, 914–922 (2008).
Wu, J. et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 46, 1769–1778 (2007).
Dolganiuc, A. et al. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127, 1513–1524 (2004).
Chang, S., Dolganiuc, A. & Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 82, 479–487 (2007).
Yang, R. et al. Papillomavirus-like particles stimulate murine bone marrow-derived dendritic cells to produce α interferon and TH1 immune responses via MyD88. J. Virol. 78, 11152–11160 (2004).
Ferrero, R. L. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol. Immunol. 42, 879–885 (2005).
Uno, K. Toll-like receptor (TLR) 2 induced through TLR4 signaling initiated by Helicobacter pylori cooperatively amplifies iNOS induction in gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G1004–G1012 (2007).
Pidgeon, G. P. et al. The role of endotoxin/lipopolysaccharide in surgically induced tumour growth in a murine model of metastatic disease. Br. J. Cancer 81, 1311–1317 (1999).
Harmey, J. H. et al. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int. J. Cancer 101, 415–422 (2002).
Luo, J. L., Maeda, S., Hsu, L. C., Yagita, H. & Karin, M. Inhibition of NF-κB in cancer cells converts inflammation- induced tumor growth mediated by TNFα to TRAIL-mediated tumor regression. Cancer Cell 6, 297–305 (2004).
Huang, B. et al. Listeria monocytogenes promotes tumor growth via tumor cell Toll-like receptor 2 signaling. Cancer Res. 67, 4346–4352 (2007).
Jego, G., Bataille, R., Geffroy-Luseau, A., Descamps, G. & Pellat-Deceunynck, C. Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia 20, 1130–1137 (2006).
Bohnhorst, J. et al. Toll-like receptors mediate proliferation and survival of multiple myeloma cells. Leukemia 20, 1138–1144 (2006).
Huang, B. et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 65, 5009–5014 (2005).
Maeda, S., Kamata, H., Luo, J. L., Leffert, H. & Karin, M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121, 977–990 (2005).
Naugler, W. E. et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317, 121–124 (2007).
Rakoff-Nahoum, S. & Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317, 124–127 (2007).
Kinzler, K. W. & Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 87, 159–170 (1996).
Oshima, M. et al. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87, 803–809 (1996).
Chulada, P. C. et al. Genetic disruption of Ptgs-1, as well as of Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 60, 4705–4708 (2000).
Wilson, C. L., Heppner, K. J., Labosky, P. A., Hogan, B. L. & Matrisian, L. M. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc. Natl Acad. Sci. USA 94, 1402–1407 (1997).
Hong, K. H., Bonventre, J. C., O'Leary, E., Bonventre, J. V. & Lander, E. S. Deletion of cytosolic phospholipase A2 suppresses ApcMin-induced tumorigenesis. Proc. Natl Acad. Sci. USA 98, 3935–3939 (2001).
Swann, J. B. et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc. Natl Acad. Sci. USA 105, 652–656 (2008).
Rakoff-Nahoum, S., Hao, L. & Medzhitov, R. Role of Toll-like receptors in spontaneous commensal-dependent colitis. Immunity 25, 319–329 (2006).
Okazaki, I. Role of AID in tumorigenesis. Adv. Immunol. 94, 245–273 (2007).
Xu, Z. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit. Rev. Immunol. 27, 367–397 (2007).
Bucala, R. Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity 26, 281–285 (2007).
Phan, R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639 (2004).
Li, M. et al. An essential role of the NF-κB/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J. Immunol. 166, 7128–7135 (2001).
Wang, J. H. Endotoxin/lipopolysaccharide activates NF-κB and enhances tumor cell adhesion and invasion through a β1 integrin-dependent mechanism. J. Immunol. 170, 795–804 (2003).
Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 (2007).
Shchors, K. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1β. Genes Dev. 20, 2527–2538 (2006).
Coussens, L. M. et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 13, 1382–1397 (1999).
Soucek, L. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nature Med. 13, 1211–1218 (2007).
Kitano, H. Cancer as a robust system: implications for anticancer therapy. Nature Rev. Cancer 4, 227–235 (2004).
Dvorak, H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
El-Omar, E. M., Ng, M. T. & Hold, G. L. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene 27, 244–252 (2008).
Apetoh, L., Tesniere, A., Ghiringhelli, F., Kroemer, G. & Zitvogel, L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. 68, 4026–4030 (2008).
Achyut, B. R., Ghoshal, U. C., Moorchung, N. & Mittal, B. Association of Toll-like receptor-4 (Asp299Gly and Thr399Ileu) gene polymorphisms with gastritis and precancerous lesions. Hum. Immunol. 68, 901–907 (2007).
He, J. F. et al. Genetic polymorphisms of TLR3 are associated with nasopharyngeal carcinoma risk in Cantonese population. BMC Cancer 7, 194 (2007).
Zhou, X. X. et al. Sequence variants in toll-like receptor 10 are associated with nasopharyngeal carcinoma risk. Cancer Epidemiol. Biomarkers Prev. 15, 862–866 (2006).
Chen, Y. C. et al. Sequence variants of Toll-like receptor 4 and susceptibility to prostate cancer. Cancer Res. 65, 11771–11778 (2005).
Zheng, S. L. et al. Sequence variants of Toll-like receptor 4 are associated with prostate cancer risk: results from the CAncer Prostate in Sweden Study. Cancer Res. 64, 2918–2922 (2004).
Sun, J. et al. Sequence variants in Toll-like receptor gene cluster (TLR6–TLR1–TLR10) and prostate cancer risk. J. Natl Cancer Inst. 97, 525–532 (2005).
Chen, Y. C., Giovannucci, E., Kraft, P., Lazarus, R. & Hunter, D. J. Association between Toll-like receptor gene cluster (TLR6, TLR1, and TLR10) and prostate cancer. Cancer Epidemiol. Biomarkers Prev. 16, 1982–1989 (2007).
Nieters, A., Beckmann, L., Deeg, E. & Becker, N. Gene polymorphisms in Toll-like receptors, interleukin-10, and interleukin-10 receptor α and lymphoma risk. Genes Immun. 7, 615–624 (2006).
Forrest, M. S. et al. Polymorphisms in innate immunity genes and risk of non-Hodgkin lymphoma. Br. J. Haematol. 134, 180–183 (2006).
Ohara, T., Morishita, T., Suzuki, H. & Hibi, T. Heterozygous Thr 135 Ala polymorphism at leucine-rich repeat (LRR) in genomic DNA of Toll-like receptor 4 in patients with poorly-differentiated gastric adenocarcinomas. Int. J. Mol. Med. 18, 59–63 (2006).
Boraska Jelavic, T. et al. Microsatelite GT polymorphism in the toll-like receptor 2 is associated with colorectal cancer. Clin. Genet. 70, 156–160 (2006).
Song, C., Chen, L. Z., Zhang, R. H., Yu, X. J. & Zeng, Y. X. Functional variant in the 3′-untranslated region of Toll-like receptor 4 is associated with nasopharyngeal carcinoma risk. Cancer Biol. Ther. 5, 1285–1291 (2006).
Hold, G. L. et al. A functional polymorphism of Toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology 132, 905–912 (2007).
Tahara, T. et al. Toll-like receptor 2 – 196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci. 98, 1790–1794 (2007).
Cerhan, J. R. et al. Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood 110, 4455–4463 (2007).
Author information
Authors and Affiliations
Corresponding author
Related links
Related links
DATABASES
National Cancer Institute Drug Dictionary
Pathway interaction Database
Rights and permissions
About this article
Cite this article
Rakoff-Nahoum, S., Medzhitov, R. Toll-like receptors and cancer. Nat Rev Cancer 9, 57–63 (2009). https://doi.org/10.1038/nrc2541
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrc2541
This article is cited by
-
Promoter methylation levels of microRNA-124 in non-neoplastic rectal mucosa as a potential biomarker for ulcerative colitis-associated colorectal cancer in pediatric-onset patients
Surgery Today (2024)
-
Porphyromonas gingivalis Lipopolysaccharide Damages Mucosal Barrier to Promote Gastritis-Associated Carcinogenesis
Digestive Diseases and Sciences (2024)
-
Mechanisms by which the intestinal microbiota affects gastrointestinal tumours and therapeutic effects
Molecular Biomedicine (2023)
-
Herbal melanin modulates PGE2 and IL-6 gastroprotective markers through COX-2 and TLR4 signaling in the gastric cancer cell line AGS
BMC Complementary Medicine and Therapies (2023)
-
Immunological characteristics of immunogenic cell death genes and malignant progression driving roles of TLR4 in anaplastic thyroid carcinoma
BMC Cancer (2023)
