
Abstract
Chorea is a common movement disorder that can be caused by a large variety of structural, neurochemical (including pharmacologic), or metabolic disturbances to basal ganglia function, indicating the vulnerability of this brain region. The diagnosis is rarely indicated by the simple phenotypic appearance of chorea, and can be challenging, with many patients remaining undiagnosed. Clues to diagnosis may be found in the patient’s family or medical history, on neurologic examination, or upon laboratory testing and neuroimaging. Increasingly, advances in genetic medicine are identifying new disorders and expanding the phenotype of recognized conditions. Although most therapies at present are supportive, correct diagnosis is essential for appropriate genetic counseling, and ultimately, for future molecular therapies.
Similar content being viewed by others

References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Kenney C, Powell S, Jankovic J. Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov Disord. 2007;22:127–30.
Rosenblatt A, Liang KY, Zhou H, et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. 2006;66:1016–20.
Wexler NS, Lorimer J, Porter J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. 2004;101:3498–503.
Aziz NA, Jurgens CK, Landwehrmeyer GB, et al. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology. 2009;73:1280–5.
Holmes SE, O'Hearn E, Rosenblatt A, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–8.
Stevanin G, Fujigasaki H, Lebre AS, et al. Huntington's disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003;126:1599–603.
Margolis RL, Holmes SE, Rosenblatt A, et al. Huntington’s Disease-like 2 (HDL2) in North America and Japan. Ann Neurol. 2004;56:670–4.
Walker RH, Jankovic J, O'Hearn E, Margolis RL. Phenotypic features of huntington disease-like 2. Mov Disord. 2003;18:1527–30.
Walker RH, Rasmussen A, Rudnicki D, et al. Huntington’s Disease-like 2 can present as chorea-acanthocytosis. Neurology. 2003;61:1002–4.
• Durr A: Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010, 9: 885–894. This is a nice summary of clinical and genetic features autosomal-dominant ataxias.
Namekawa M, Takiyama Y, Ando Y, et al. Choreiform movements in spinocerebellar ataxia type 1. J Neurol Sci. 2001;187:103–6.
Geschwind DH, Perlman S, Figueroa CP, et al. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet. 1997;60:842–50.
Rottnek M, Riggio S, Byne W, et al. Schizophrenia in a patient with spinocerebellar ataxia 2: coincidence of two disorders or a neurodegenerative disease presenting with psychosis? Am J Psychiatry. 2008;165:964–7.
Lee WW, Kim SY, Kim JY, et al. Extrapyramidal signs are a common feature of spinocerebellar ataxia type 17. Neurology. 2009;73:1708–9.
Le Ber I, Camuzat A, Castelnovo G, et al. Prevalence of dentatorubral-pallidoluysian atrophy in a large series of white patients with cerebellar ataxia. Arch Neurol. 2003;60:1097–9.
Wardle M, Majounie E, Williams NM, et al. Dentatorubral pallidoluysian atrophy in South Wales. J Neurol Neurosurg Psychiatry. 2008;79:804–7.
Burke JR, Wingfield MS, Lewis KE, et al. The Haw River Syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African-American family. Nat Genet. 1994;7:521–4.
Mahajnah M, Inbar D, Steinmetz A, et al. Benign hereditary chorea: clinical, neuroimaging, and genetic findings. J Child Neurol. 2007;22:1231–4.
Bauer P, Kreuz FR, Burk K, et al. Mutations in TITF1 are not relevant to sporadic and familial chorea of unknown cause. Mov Disord. 2006;21:1734–7.
Asmus F, Horber V, Pohlenz J, et al. A novel TITF-1 mutation causes benign hereditary chorea with response to levodopa. Neurology. 2005;64:1952–4.
Krude H, Schutz B, Biebermann H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002;109:475–80.
Curtis AR, Fey C, Morris CM, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–4.
Crompton DE, Chinnery PF, Bates D, et al. Spectrum of movement disorders in neuroferritinopathy. Mov Disord. 2004;20:95–9.
Kubota A, Hida A, Ichikawa Y, et al. A novel ferritin light chain gene mutation in a Japanese family with neuroferritinopathy: description of clinical features and implications for genotype-phenotype correlations. Mov Disord. 2009;24:441–5.
Suls A, Dedeken P, Goffin K, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. 2008;131:1831–44.
• Leen WG, Klepper J, Verbeek MM, et al.: Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133: 655–670. This is a recent update on this treatable disorder.
Geschwind DH, Loginov M, Stern JM. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification (Fahr disease). Am J Hum Genet. 1999;65:764–72.
Oliveira JR, Spiteri E, Sobrido MJ, et al. Genetic heterogeneity in familial idiopathic basal ganglia calcification (Fahr disease). Neurology. 2004;63:2165–7.
Wszolek ZK, Baba Y, Mackenzie IR, et al. Autosomal dominant dystonia-plus with cerebral calcifications. Neurology. 2006;67:620–5.
Younes-Mhenni S, Thobois S, Streichenberger N, et al. Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (Melas) associated with a Fahr disease and cerebellar calcifications. Rev Med Interne. 2002;23:1027–9.
Kovacs GG, Murrell JR, Horvath S, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–7.
Gamez J, Corbera-Bellalta M, Mila M, et al. Chorea-ballism associated with familial amyotrophic lateral sclerosis. A clinical, genetic, and neuropathological study. Mov Disord. 2008;23:434–8.
Pradat PF, Salachas F, Lacomblez L, et al. Association of chorea and motor neuron disease. Mov Disord. 2002;17:419–20.
Nielsen TR, Bruhn P, Nielsen JE, Hjermind LE. Behavioral variant of frontotemporal dementia mimicking Huntington's disease. Int Psychogeriatr. 2010;22:674–7.
Machado A, Fen CH, Mitiko DM, et al. Neurological manifestations in Wilson's disease: report of 119 cases. Mov Disord. 2006;21:2192–6.
Zhu D, Burke C, Leslie A, Nicholson GA. Friedreich's ataxia with chorea and myoclonus caused by a compound heterozygosity for a novel deletion and the trinucleotide GAA expansion. Mov Disord. 2002;17:585–9.
Spacey SD, Szczygielski BI, Young SP, et al. Malaysian siblings with friedreich ataxia and chorea: a novel deletion in the frataxin gene. Can J Neurol Sci. 2004;31:383–6.
• Verhagen MMM, Abdo WF, Willemsen MAAP, et al.: Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology 2009, 73: 430–437. This article reports on features of this disorder in previously misdiagnosed adults.
Rampoldi L, Danek A, Monaco AP. Clinical features and molecular bases of neuroacanthocytosis. J Mol Med. 2002;80:475–91.
Storch A, Kornhass M, Schwarz J. Testing for acanthocytosis—a prospective reader-blinded study in movement disorder patients. J Neurol. 2005;252:84–90.
Rampoldi L, Dobson-Stone C, Rubio JP, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28:119–20.
Dobson-Stone C, Velayos-Baeza A, Filippone LA, et al. Chorein detection for the diagnosis of chorea-acanthocytosis. Ann Neurol. 2004;56:299–302.
Velayos-Baeza A, Holinski-Feder E, Nietzel B, et al.: Chorea-acanthocytosis genotype in Critchley's original Kentucky neuroacanthocytosis kindred. Arch Neurol. in press.
Mubaidin A, Roberts E, Hampshire D, et al. Karak syndrome: a novel degenerative disorder of the basal ganglia and cerebellum. J Med Genet. 2003;40:543–6.
Miyajima H. Aceruloplasminemia, an iron metabolic disorder. Neuropathology. 2003;23:345–50.
McNeill A, Pandolfo M, Kuhn J, et al. The neurological presentation of ceruloplasmin gene mutations. Eur Neurol. 2008;60:200–5.
Friedman JR, Thiele EA, Wang D, et al. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov Disord. 2006;21:241–5.
Sethi KD, Ray R, Roesel RA, et al. Adult-onset chorea and dementia with propionic acidemia. Neurology. 1989;39:1343–5.
Gascon GG, Ozand PT, Brismar J. Movement disorders in childhood organic acidurias. Clinical, neuroimaging, and biochemical correlations. Brain Dev. 1994;16(Suppl):94–103.
Hall DA, Ringel SP. Adult nonketotic hyperglycinemia (NKH) crisis presenting as severe chorea and encephalopathy. Mov Disord. 2004;19:485–6.
Morrison PF, Sankar R, Shields WD. Valproate-induced chorea and encephalopathy in atypical nonketotic hyperglycinemia. Pediatr Neurol. 2006;35:356–8.
Mellick G, Price L, Boyle R. Late-onset presentation of pyruvate dehydrogenase deficiency. Mov Disord. 2004;19:727–9.
Brown RM, Head RA, Morris AA, et al. Pyruvate dehydrogenase E3 binding protein (protein X) deficiency. Dev Med Child Neurol. 2006;48:756–60.
Shulman LM, Lang AE, Jankovic J, et al. Case 1, 1995: psychosis, dementia, chorea, ataxia, and supranuclear gaze dysfunction. Mov Disord. 1995;10:257–62.
Oates CE, Bosch EP, Hart MN. Movement disorders associated with chronic GM2 gangliosidosis. Case report and review of the literature. Eur Neurol. 1986;25:154–9.
Danek A, Rubio JP, Rampoldi L, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol. 2001;50:755–64.
Hewer E, Danek A, Schoser BG, et al. McLeod myopathy revisited—more neurogenic and less benign. Brain. 2007;130:3285–96.
Evidente VG, Advincula J, Esteban R, et al. Phenomenology of “Lubag” or X-linked dystonia-parkinsonism. Mov Disord. 2002;17:1271–7.
Factor SA, Barron KD. Mosaic pattern of gliosis in the neostratum of a North American man with craniocervical dystonia and parkinsonism. Mov Disord. 1997;12:783–9.
Gibb WR, Kilford L, Marsden CD. Severe generalised dystonia associated with a mosaic pattern of striatal gliosis. Mov Disord. 1992;7(3):217–23.
Goldenberg PC, Steiner RD, Merkens LS, et al. Remarkable improvement in adult Leigh syndrome with partial cytochrome c oxidase deficiency. Neurology. 2003;60:865–8.
Crimi M, Galbiati S, Moroni I, et al. A missense mutation in the mitochondrial ND5 gene associated with a Leigh-MELAS overlap syndrome. Neurology. 2003;60:1857–61.
Caer M, Viala K, Levy R, et al. Adult-onset chorea and mitochondrial cytopathy. Mov Disord. 2005;20:490–2.
Morimoto N, Nagano I, Deguchi K, et al. Leber hereditary optic neuropathy with chorea and dementia resembling Huntington disease. Neurology. 2004;63:2451–2.
Lee BC, Hwang SH, Chang GY. Hemiballismus-hemichorea in older diabetic women: a clinical syndrome with MRI correlation. Neurology. 1999;52:646–8.
Ahlskog JE, Nishino H, Evidente VG, et al. Persistent chorea triggered by hyperglycemic crisis in diabetics. Mov Disord. 2001;16:890–8.
Shyambabu C, Sinha S, Taly AB, et al. Serum vitamin B12 deficiency and hyperhomocystinemia: a reversible cause of acute chorea, cerebellar ataxia in an adult with cerebral ischemia. J Neurol Sci. 2008;273:152–4.
Pacchetti C, Cristina S, Nappi G. Reversible chorea and focal dystonia in vitamin B12 deficiency. N Engl J Med. 2002;347:295.
Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med. 2003;9:914–20.
Bowen J, Mitchell T, Pearce R, Quinn N. Chorea in new variant Creutzfeldt-Jacob disease. Mov Disord. 2000;15:1284–5.
McKee D, Talbot P. Chorea as a presenting feature of variant Creutzfeldt-Jakob disease. Mov Disord. 2003;18:837–8.
Passarin MG, Alessandrini F, Nicolini GG, et al. Reversible choreoathetosis as the early onset of HIV-encephalopathy. Neurol Sci. 2005;26:55–6.
Sporer B, Linke R, Seelos K, et al. HIV-induced chorea: evidence for basal ganglia dysregulation by SPECT. J Neurol. 2005;252:356–8.
Ozben S, Erol C, Ozer F, Tiras R. Chorea as the presenting feature of neurosyphilis. Neurol India. 2009;57:347–9.
Font J, Cervera R, Espinosa G, et al. Systemic lupus erythematosus (SLE) in childhood: analysis of clinical and immunological findings in 34 patients and comparison with SLE characteristics in adults. Ann Rheum Dis. 1998;57:456–9.
Watanabe T, Onda H. Hemichorea with antiphospholipid antibodies in a patient with lupus nephritis. Pediatr Nephrol. 2004;19:451–3.
Venegas FP, Sinning M, Miranda M. Primary Sjogren’s syndrome presenting as a generalized chorea. Parkinsonism Relat Disord. 2005;11:193–4.
Ciubotaru CR, Esfahani F, Benedict RH, et al. Chorea and rapidly progressive subcortical dementia in antiphospholipid syndrome. J Clin Rheumatol. 2002;8:332–9.
Kumar H, Masiowski P, Jog M. Chorea in the elderly with mutation positive polycythemia vera: a case report. Can J Neurol Sci. 2009;36:370–2.
Pereira AC, Edwards MJ, Buttery PC, et al. Choreic syndrome and coeliac disease: a hitherto unrecognised association. Mov Disord. 2004;19:478–82.
Honnorat J, Cartalat-Carel S, Ricard D, et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry. 2009;80:412–6.
Dorban S, Gille M, Kessler R, et al. Chorea-athetosis in the anti-Hu syndrome. Rev Neurol (Paris). 2004;160:126–9.
Krolak-Salmon P, Androdias G, Meyronet D, et al. Slow evolution of cerebellar degeneration and chorea in a man with anti-Yo antibodies. Eur J Neurol. 2006;13:307–8.
• Vincent A, Bien CG: Anti-NMDA-receptor encephalitis: a cause of psychiatric, seizure, and movement disorders in young adults. Lancet Neurol 2008, 7: 1074–1075. This is a review of this recently recognized, but not uncommon, condition.
Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
Surie S, Tijssen MA, Biervliet JD, et al. Chorea in adults following pulmonary endarterectomy. Mov Disord. 2010;25:1101–4.
Passarin MG, Romito S, Avesani M, et al. Late-onset choreoathetotic syndrome following heart surgery. Neurol Sci. 2010;31:95–7.
Suchowersky O, Muthipeedika J. A case of late-onset chorea. Nat Clin Pract Neurol. 2005;1:113–6.
Gironell A, de Molina RM, Sancho G, Kulisevsky J: Chorea induced by a luteinizing hormone-releasing hormone analog. J Neurol. 2008;255:1264–5.
Bota DA, Dafer RM. Acute methotrexate neurotoxicity with choreiform movements and focal neurological deficits: a case report. South Med J. 2009;102:1071–4.
Necioglu OD, Yldrmak Y, Kenangil G, et al. Intrathecal methotrexate-induced acute chorea. J Pediatr Hematol Oncol. 2009;31:57–8.
van der Plas AA, van Rijn MA, van Hilten JJ. Baclofen-induced chorea in complex regional pain syndrome-related dystonia. Mov Disord. 2010;25:959–60.
Weiner WJ, Nausieda PA, Klawans HL. Methylphenidate-induced chorea: case report and pharmacologic implications. Neurology. 1978;28:1041–4.
Thiriaux A, de St Martin A, Vercueil L, et al. Co-occurrence of infantile epileptic seizures and childhood paroxysmal choreoathetosis in one family: clinical, EEG, and SPECT characterization of episodic events. Mov Disord. 2002;17:98–104.
Rainier S, Thomas D, Tokarz D, et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch Neurol. 2004;61:1025–9.
Gancher ST, Nutt JG. Autosomal dominant episodic ataxia: a heterogeneous syndrome. Mov Disord. 1986;1:239–53.
Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–40.
Biolsi B, Cif L, Fertit HE, et al. Long-term follow-up of Huntington disease treated by bilateral deep brain stimulation of the internal globus pallidus. J Neurosurg. 2008;109:130–2.
Kang GA, Heath S, Rothlind J, Starr PA: Long-term follow-up of pallidal deep brain stimulation in two cases of Huntington's disease. J Neurol Neurosurg Psychiatry 2010
Kaufman CB, Mink JW, Schwalb JM. Bilateral deep brain stimulation for treatment of medically refractory paroxysmal nonkinesigenic dyskinesia. J Neurosurg. 2010;112:847–50.
Cicchetti F, Saporta S, Hauser RA, et al. Neural transplants in patients with Huntington's disease undergo disease-like neuronal degeneration. Proc Natl Acad Sci U S A. 2009;106:12483–8.
Bachoud-Levi AC, Gaura V, Brugieres P, et al. Effect of fetal neural transplants in patients with Huntington's disease 6 years after surgery: a long-term follow-up study. Lancet Neurol. 2006;5:303–9.
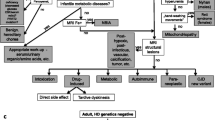
Walker RH. Introduction: an approach to the patient with chorea. In: Walker RH, editor. The differential diagnosis of chorea. Oxford: Oxford University Press; 2010.
Disclosure
Conflicts of interest: R.H. Walker: has received honoraria from Bioavail, and has received payment from Scienta and Intellyst.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Walker, R.H. Differential Diagnosis of Chorea. Curr Neurol Neurosci Rep 11, 385–395 (2011). https://doi.org/10.1007/s11910-011-0202-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-011-0202-2