
Purpose
Fusogenic liposomes (FLs) are unique delivery vehicles capable of introducing their contents directly into the cytoplasm with the aid of envelope glycoproteins of Sendai virus (SeV). The objective of this study was to evaluate the potential of FL to improve the mucosal absorption of insulin from rat intestinal membranes.
Method
The FLs containing insulin were prepared by fusing insulin-loaded liposomes with inactivated SeV particles and were administered directly into the ileal, the colonic, and the rectal loops (10 IU/kg).
Results
The FL successfully enhanced the insulin absorption and induced a significant hypoglycemic effect following the colonic and the rectal administration without detectable mucosal damage. This enhancing effect of insulin absorption was further improved by increasing the amount of insulin loaded in the FL and by coencapsulating insulin-degrading enzyme inhibitor. In contrast, the insulin absorption was not increased by the ileal administration of FL because the mucous/glycocalyx layers overlaid on the ileal epithelium impede the fusion of FL to the intestinal mucosa.
Conclusion
Our results indicated that FL is a useful carrier for improving the absorption of poorly absorbable drugs, such as insulin, via the intestinal tract.
Similar content being viewed by others


Introduction
The major problems existing in developing intestinal delivery systems for peptide and protein drugs are the inactivation by the proteolytic enzymes in the gastrointestinal tract and limitation of its membrane permeability. Various approaches, including absorption enhancers (1,2), enzyme inhibitors (1,3), and chemical modification (4,5), have been studied to overcome these problems. In addition to such strategies, liposomes have been given much attention because the incorporated protein and peptide drugs could be protected from gastrointestinal digestion and immune recognition (6), and many studies have demonstrated that liposomes can improve the enteral absorption of peptide and protein drugs (6,7). However, these conventional liposomes have not shown sufficient bioavailability when administered by the oral route.
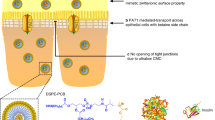
Fusogenic liposomes (FLs) are unique delivery vehicles equipped with the envelope glycoprotein of Sendai virus (SeV). The FLs are prepared by fusing conventional liposomes with inactivated SeV particles and can deliver encapsulated contents directly and efficiently into the cytoplasm through membrane fusion, with the same mechanism as SeV infection (8–10). So far, these SeV-mediated drug delivery systems have been used to successfully introduce many kinds of macromolecules, such as DNA, proteins, and nanocarriers, into animal cells (8–10).
The objective of this study was to evaluate the potential of FL to improve the mucosal absorption of peptide and protein drugs. Although it has been reported that FL had a wide range of target cells, their applicability to the intestinal membrane has not yet been established. Therefore, we first investigated the infectivity of SeV to the various intestinal mucosae. The mucous/glycocalyx layers have been known to envelop the surface of the intestinal epithelium and are regarded as an undesirable diffusion barrier that impedes access to absorptive cells of macromolecules and particles (11,12). As this layer may also impede the accessibility of FL to the intestinal epithelium, we examined the effect of removing the mucous/glycocalyx layers by hyaluronidase pretreatment on membrane fusion. In a previous study, we proved that the mucous/glycocalyx layers were successfully diminished without causing detectable cellular damage by hyaluronidase pretreatment (13,14). We then examined the absorption of peptide drugs from various intestinal mucosae with the aid of FL, using insulin as a model peptide drug. One of the limiting factors for improving insulin absorption from the intestinal epithelium may be the degradation of insulin in the cytosol by insulin-degrading enzyme (IDE) (15,16). IDE is the neutral thiol metalloproteinase involved in the intracellular metabolism of insulin in hepatocytes, adipocytes, kidney, muscle cells, intestinal tissue, and other cells (15–17). Therefore, we investigated whether the coadministration of IDE inhibitor would further increase the absorption enhancement of insulin by the intestinal administration of FL. Finally, to insure the safety of FL as an intestinal delivery carrier, the intestinal damage was evaluated by lactate dehydrogenase (LDH) leakage.
Materials and Methods
Materials
Recombinant human insulin (26.0 IU/mg) was purchased from Wako Pure Chemical Industries, Co., Ltd. (Osaka, Japan). Lyophilized hyaluronidase (EC 3.2.1.35; Type IV-S from bovine testes; MW = 56 kDa, 1370 IU/mg solid), sodium taurodeoxycholate, and p-chloromercuribenzoate (PCMB) were purchased from Sigma-Aldrich Chemical Co., Ltd. (St. Louis, MO, USA). Purified egg yolk phosphatidylcholine (PC), l-α-dimyristoyl phosphatidylglycerol (PG), and cholesterol (Chol) were obtained from Nippon Oil and Fats Co., Ltd. (Tokyo, Japan). Polycarbonate membranes (pore size 0.2 μm, Anodisc) were obtained from Whatman PLC (Kent, UK). SeV (Z strain) was prepared in embryonated chicken eggs as described previously (18). All other chemicals were used of reagent grade without further purification.
Fusion Study of SeV
This research complied with the regulations of the Committee on Ethics in the Care and Use of Laboratory Animals of National Institute of Advanced Industrial Science and Technology, according to the institutional regulations of recombinant DNA experiments. Expression of the green fluorescent protein (GFP) gene carried by recombinant SeV (GFP-SeV) was used to probe the SeV infection (e.g., delivery of the viral genome through membrane fusion), and UV-inactivated virus was used as a negative control. Male Sprague-Dawley rats (180–220 g) were purchased from Tokyo Laboratory Amimals Science Co., Ltd. (Tokyo, Japan). Animals were housed in rooms with temperature controlled between 23 ± 1°C and 55 ± 5% relative humidity and were allowed free access to water and food during acclimatization, but they were fasted for 24 h before experiments. Following anesthetization by the intraperitoneal injection of sodium pentobarbital (50 mg/kg; Dainippon Pharmaceutical Co., Ltd., Osaka, Japan), the rats were restrained in a supine position on a thermostatically controlled board at 37°C and maintained body temperature. The ileum, the colon, and the rectum were exposed following a small midline incision carefully made in the abdomen, and each segment was cannulated at both ends using polypropylene tubings (4-mm o.d., 2-mm i.d., Saint-Gobain Norton Co., Ltd., Nagano, Japan). These were securely ligated to prevent fluid loss and, subsequently, carefully returned to their original location inside the peritoneal cavity. To wash away the intestinal content, phosphate-buffered saline (PBS; pH 7.4) at 37°C was singly circulated through the cannula at 5 ml/min for 4 min using a peristaltic pump (PSK-51, Nikkiso Co., Ltd., Tokyo, Japan). Subsequently, the intestinal segments were exposed to 1.0 ml of PBS as a control and hyaluronidase solution dissolved in PBS (1.92 × 105 U/ml) for 30 min. After exposure, the segments were carefully rinsed with 20 ml of PBS at 37°C, and the segments were tightly closed following the removal of the cannulation tubing. The SeV suspensions were directly administered to the intestinal segments, and the abdomen was sutured. At 16 h following administration, each segment was removed, and tissue specimens were prepared. The specimens treated with the tissue-embedding medium OCT compound (Tissue-tek®, Sakura Finetek, Inc., Torrance, CA, USA) were frozen in acetone, and 20-μm transverse frozen sections were made in a cryostat (Leica CM 1850, Leica Microsystems, Co., Ltd., Heidelberg, Germany) at −20°C. Sections were placed on slides and dried at room temperature. To prevent the bleaching of the fluorescence during microscopic examination, the sections were mounted in antifading reagents (Aqua-Poly, Polysciences, Inc., Warrington, PA, USA). Expression of the GFP induced by GFP-SeV was determined by confocal laser scanning microscopy (Radiance 2100, Bio-Rad Laboratories, Hercules, CA, USA). All photographs were taken with ×100 magnification.
Preparation and Characterization of FL Containing Insulin
Multilamellar liposomes were prepared by the freezing–thawing method using 120-mg lipids (PC/PG/Chol = 4:1:5 molar ratio). The lipid mixtures were dissolved in a small amount of benzene/methanol mixture (95:5%, v/v), and the solvent was rotary evaporated to obtain a thin lipid film. The liposome suspension was prepared by dispersing the dried lipid film with a given amount of insulin solution (either 10 or 30 mg/ml) with or without IDE inhibitor PCMB (1 mg/ml) using a vortex mixer. The liposomes were frozen rapidly in deep freezer and left to thaw at 37°C. After five cycles of freezing and thawing, unilamellar liposomes were prepared by extrusion through a 0.2-μm polycarbonate membrane and were separated from unencapsulated drugs by ultracentrifugation (40,000 rpm, 90 min). Unilamellar FLs were prepared as described elsewhere (8–10). Briefly, unilamellar liposomes were mixed with SeV and incubated at 37°C for 2 h with shaking. The FL was separated from free liposomes and SeV by sucrose density-gradient centrifugation (40,000 rpm, 90min). The amount of insulin and PCMB loaded into liposomes was determined by high-performance liquid chromatography (HPLC). The HPLC was composed of a pump (LC-10AS, Shimadzu Co. Ltd., Kyoto, Japan), a UV detector (SPD-10A, Shimadzu), a system controller (SCL-10A), an autoinjector (SIL-10A, Shimadzu), an integrator (C-R3A, Shimadzu), and a GL-PACK Nucleosil 100-5C18 column (150 × 4.6 mm i.d.). The mobile phase was a mixture of acetonitrile/0.1% trifluoroacetic acid/NaCl = 31:69:0.58% (v/v/w). The flow rate was 1.0 ml/min, and UV detection was performed at a wavelength of 220 nm. The encapsulation efficiency of insulin and PCMB into FL was calculated based on the initial and loading amount of drugs. In this study, 3.3 and 13.0% of insulin and PCMB in the initial solution was loaded into FL, respectively.
In Situ Absorption Study
This research complied with the regulations of the Committee on Ethics in the Care and Use of Laboratory Animals of Hoshi University. In situ absorption study was performed in the same in situ system as described above, except that the hyaluronidase treatment was omitted. The ileum, the colon, and the rectum were treated with PBS at 37°C to wash away the intestinal content. Rats were further left on the board at 37°C for 1 h to be recovered from the elevated blood glucose levels because of the surgical operations describe above. Following 1 h of rest, the insulin solution or each liposome was directly administered into the ileal, the colonic, and the rectal loops. The dose of all samples was fixed at 5 and 10 IU/kg body weight. During the experiment, a 0.2-ml aliquot of blood was collected from the jugular vein at 0, 5, 10, 15, 30, 60, 120, 180, and 240 min following administration. To calculate the efficacy of enteral insulin administration relative to subcutaneous administration, insulin solutions were administered subcutaneously. Insulin solution was prepared by dissolving an appropriate amount of crystalline human insulin in PBS. The insulin subcutaneous dose was 1 IU/kg body weight. Blood samples (0.2 ml) were collected from the jugular vein at the injection before, 5, 10, 15, 30, 60, 120, 180, and 240 min following administration. To set the same physical conditions on the rats, the animals receiving s.c. injection were given the same surgical operation as the in situ absorption study. The plasma was separated by centrifugation at 13,000 rpm for 1 min and was kept in a freezer until insulin concentration analysis. The plasma insulin concentrations were determined by enzyme immunoassay using a microplate luminometer (Mithras LB940, Beltold Japan Co., Ltd., Osaka, Japan). Blood glucose levels were determined using a glucose meter (Novo Assist Plus, Novo Nordisk Pharma Ltd., Tokyo, Japan).
The total area under the insulin concentration curve (AUC) from time 0 to 240 min was estimated from the sum of successive trapezoids between each data point. The bioavailability was calculated to the s.c. injection as described above. The plasma peak level (C max) and the time taken to reach the plasma peak level were determined from the plasma insulin level–time curves. The area above the blood glucose levels–time curve was calculated by a trapezoidal rule. The pharmacological bioavailability (PA) was calculated relative to the insulin s.c. injection, as described above.
Biochemical Evaluation of Intestinal Damage
The colonic loop from the pretreated segment following the in situ experiments as describe above was used here. The colonic segment was treated with PBS (20 ml) at 37°C to wash away the intestinal content. PBS (control), 1% (w/v) sodium taurodeoxycholate (positive control), or FL was administered to the colon and incubated in the segments for 2 h. Then, the colonic loop was washed with 1.0 ml of PBS, and the intestinal fluid was collected. The concentration of LDH in the fluid was determined using a LDH-Test Wako (Wako Pure Chemical Industries, Co., Ltd.).
Statistical Analysis
The results were expressed as the mean ± SD. For group comparison, an analysis of variance (ANOVA) with a one-way layout was applied. Significant differences in the mean values were evaluated using Student's t test. Differences were considered to be significant when the p value was less than 0.05.
Results
Fusion of SeV to the Various Intestinal Mucosae
Figure 1 shows GFP expression in the various intestinal membranes following the administration of GFP-SeV or UV-inactivated GFP-SeV. GFP expression was clearly detected in each mucosal membrane after treatment with GFP-SeV, but not in those treated with UV-inactivated virus, indicating that the GFP gene was successfully delivered into each mucosal segment through membrane fusion. In the ileal segments, hyaluronidase pretreatment significantly enhanced the GFP expression (Fig. 1D) compared with those untreated (Fig. 1A), whereas it did not affect the GFP expression in the colon (Fig. 1E vs. B) and the rectum (Fig. 1C vs. F). These results suggest that the pre-epithelial mucous/glycocalyx layers constitute a barrier for the fusion of SeV with the ileal membrane, but not with the colonic and the rectal membranes.

Green fluorescent protein (GFP) expression in the ileal (A, D, G), colonic (B, E, H), and rectal (C, F, I) mucosa treated with GFP-Sendai virus (SeV; A–F) or UV-inactivated GFP-SeV (G–I). Each intestinal segment was pretreated with phosphate-buffered saline (PBS; A–C, G–I) and hyaluronidase (D–F). Each picture was taken by ×100 magnification.
Intestinal Absorption of Insulin Following the in Situ Administration of Insulin-Loaded FL
Figure 2 shows the time course of the plasma insulin and blood glucose levels following in situ administration of insulin-loaded FL at a dose of 10 IU/kg into the colonic segments. No apparent insulin absorption and hypoglycemic effect were observed in the insulin solution. Similarly, insulin-loaded liposomes did not enhance insulin absorption, although a slight hypoglycemic effect was observed. In contrast, a marked increase in the plasma insulin levels accompanied by a decrease in the blood glucose levels was observed following the in situ administration of insulin-loaded FL into the colonic segments.

Plasma insulin (A) and blood glucose (B) levels vs. time profiles following colonic administration of insulin-loaded fusogenic liposome (FL). Each value represents the mean ± SD from n = 3–4. Open circle, insulin solution (control); closed square, insulin-loaded liposomes; closed circle, insulin-loaded FL.
Figure 3 shows the insulin absorption following FL administration into the ileal, the colonic, and the rectal segments. In the ileal segment, no apparent insulin absorption and hypoglycemic effect were observed following FL administration, whereas FL induced the dramatic insulin absorption in the colonic and the rectal segments.

Plasma insulin (A) and blood glucose (B) levels vs. time profiles following in situ administration of insulin-loaded FL into various intestinal regions. Each value represents mean ± SD from n = 3–4. Open circle, ileum; closed square, colon; closed circle, rectum.
Effect of Loading Amounts on the Insulin Colonic Absorption
Figure 4 shows the effect of the concentration of insulin encapsulated into FL on the insulin absorption from the colonic segments (A) and hypoglycemic effect (B). The FL loaded with insulin in higher concentration (30 mg/ml) showed a stronger insulin absorption enhancement effect than those loaded with insulin in lower concentration (10 mg/ml). These results imply that the average number of insulin molecules encapsulated in a FL particle is a key factor determining the efficacy of insulin delivery by FL.

Effect of insulin loading amount into FL on plasma insulin (A) and blood glucose (B) levels. Each value represents the mean ± SD from n = 3–5. Closed circle, 10-mg insulin-loaded FL; open circle, 30-mg insulin-loaded FL.
Effect of IDE Inhibitor on the Insulin Absorption Enhancement Effect by FL
Figure 5 shows the effect of PCMB coencapsulated in FL on plasma insulin (A) and on blood glucose (B) levels following the administration of insulin-loaded FL at a dose of 5 IU/kg into the colonic segments. When the insulin solution was coadministered with PCMB, the insulin absorption was not increased. Similarly, both insulin- and PCMB-loaded simple liposomes did not enhance the colonic absorption of insulin. These results indicated that PCMB did not show the absorption-enhancing effect of insulin. In contrast, the FL loaded with both insulin and PCMB together (insulin- and PCMB-loaded FL) greatly enhanced insulin absorption as compared with insulin-loaded FL. These results demonstrated that the intracellular degradation of insulin by IDE strongly affected the insulin absorption by FL.

Effect of intracolonic administration of insulin- and p-chloromercuribenzoate (PCMB)-loaded FL containing on plasma insulin (A) and blood glucose (B) levels. Each value represents the mean ± SD from n = 3–5. Closed square, insulin + PCMB solution (control); open square, insulin- and PCMB-loaded liposomes; closed circle, insulin-loaded FL; open circle, insulin- and PCMB-loaded FL.
Table I summarizes the pharmacokinetic parameters of insulin following the colonic administration of insulin-loaded FL. In this study, the pharmacological availability and relative bioavailability (BA) were calculated based on the s.c. injection of 1.0 IU/kg of insulin. Insulin-loaded FL was found to have higher values of pharmacokinetic parameters, C max, AUC, PA, and BA, than that of insulin solution and insulin-loaded liposomes. Furthermore, these parameters were markedly increased by the addition of IDE inhibitor into the insulin-loaded FL, and their PA and BA values of insulin were greatly improved, up to approximately 9 and 16%, respectively.
Biochemical Characterization of the Colonic Membrane Damage Following FL Administration
Table II shows the LDH leakage following the 2-h administration of FL to the colonic region. LDH was negligibly leaked into the colonic segment, which was similar to the leakage seen in the control group (0.39 ± 0.18 vs. 0.40 ± 0.10 U, respectively). On the other hand, LDH leakage was significantly induced by the administration of sodium taurodeoxycholate (5.13 ± 0.46 U). These data suggest that insulin-loaded FL did not induce the intestinal damage, and the absorption-enhancing effect of insulin-loaded FL was not because of a loss of membranous barrier function affected by cellular viability.
Discussion
The absorption of peptide and protein drugs from the intestine is limited because of enzymatic degradation in the gastrointestinal tract, high molecular weight, and hydrophilicity. For overcoming these limiting factors, various types of liposomes have been used to enhance intestinal insulin absorption; however, bioavailability of the peptide and protein drugs observed by liposome administration is insufficient. To improve mucosal insulin absorption by active targeting and delivery, we evaluate the potential of FL as a mucosal insulin carrier. The FL is a delivery system that can introduce the encapsulated materials into the living cells directly and efficiently through the membrane fusion (8–10). From these characteristics, it would be expected that insulin loaded into FL would be directly introduced into the intestinal epithelium, thereby increasing the intestinal insulin absorption.
As shown in Fig. 1, the SeV can deliver the GFP gene into each mucosal membrane through membrane fusion. Membrane fusion of SeV and FL was induced by two envelope glycoproteins: hemagglutinating and neuraminidase (HN) and fusion (F) proteins (19). The HN protein is involved in cell binding through attachment of the virus envelope to the cellular receptors containing sialic acid, whereas the F protein plays a major role in fusion of the envelope with the cell membrane. As sialic acid is present on the cell membrane of most cell types, including the apical surface of intestinal membrane, SeV and FL are expected to bind to the surface of intestinal epithelium and fuse with the cell membrane.
The effects of FL on the insulin intestinal absorption were dependent on the site of the administration (ileum, colon, and rectum; Fig. 3) and were consistent with the results of a fusion study of SeV to the intestinal mucosa using GFP expression as a marker (Fig. 1). As shown in Fig. 1, the fusion ability of SeV to the large intestine was higher than that to the small intestine. In addition, we found that the mucous/glycocalyx layers of the ileal mucosa affected the membrane fusion of SeV. A possible explanation of the results may be the different activities of proteolytic enzymes responsible for the degradation of the SeV envelope glycoprotein in the small and the large intestine. Pancreatic enzyme activities were much stronger in the small intestine than in the large intestine (20). It has been shown that peptide and protein drugs were degraded by pancreatic enzymes such as trypsin and chymotrypsin, which are believed to reside both in the intestinal fluids and the mucous/glycocalyx layers (21). Asano et al. (22) reported that the SeV envelope glycoproteins were easily degraded by pancreatic enzymes, such as trypsin and chymotrypsin, and lost their activities of fusion to the cell membrane. Therefore, the SeV glycoproteins may be degraded more readily in the ileal mucous/glycocalyx layers than in the colonic and the rectal mucous/glycocalyx layers, and this may explain the obvious regional differences in the insulin absorption by FL.
It was also demonstrated that the increase of the loading amount of insulin in a single FL was quite effective for facilitating the insulin absorption from the colon (Fig. 4). Accounting that the total number of FL particles is smaller in the high-loading preparation (30 mg/ml insulin) than in the low-loading preparation (10 mg/ml) because the total amount of insulin administrated was kept constant in this experiment, specific absorption of insulin per FL particle is increased over the difference in the concentration of insulin encapsulated. Based on a kinetic and quantitative analysis of the SeV fusion to the cell membrane, the number of SeV (or FL) particles capable of fusing with a single cell was limited by the number of the absorption site on the cell surface (23). Therefore, the fusion of FL to the intestinal mucosa might be already saturated even at the low concentration of FL. This may be one of the limiting factors for the use of FL to the mucosal delivery systems.
Regarding the permeation of insulin into the intestinal epithelium, Bai and Chang (15,16) reported that insulin was highly degraded by IDE in the intestinal cytoplasm, and it potentially limits the transepithelial transport of insulin. In this study, we found that the coencapsulation of PCMB as the IDE inhibitor further increased FL's enhancement effect of insulin colonic absorption (Fig. 5). This implies that insulin, which was directly introduced into the cytosol by the FL membrane fusion, was strongly degraded by the cytosolic IDE. PCMB, a sulfhydryl-modifying agent, has been used to inhibit the degradation of insulin by IDE in various tissue cytoplasms (15,16). In the present study, no absorption-enhancing effect was observed following insulin solution coadministration of PCMB and the simple liposomes loaded with both insulin and PCMB together. From these findings, it is suggested that PCMB did not have an absorption-enhancing effect, and further increasing the absorption enhancement of insulin was only due to reducing the degradation of insulin in the intestinal cytosol.
Generally, the absorption of insulin from the intestine is limited because of enzymatic degradation in the gastrointestinal tract and low permeability through the intestinal mucosa. To date, various strategies have been used to improve intestinal absorption of insulin; however, mucosal bioavailability of insulin was only improved up to 1–5% by coadministration with absorption enhancers (1) or enzyme inhibitors (1) and chemical modification (4) in the same in situ loop method used in this study. In addition, some of these approaches exhibited several negative effects such as irritation of the intestinal mucosal membrane and impairment of the membrane barrier. In this study, we observed that insulin-loaded FL successfully enhanced insulin enteral absorption, achieving approximately 9% bioavailability (Table I). The high efficacy of FL in mucosal delivery is attributed to the protection of the peptides by the liposomes and the direct introduction of insulin into the intestinal epithelial cells through the membrane fusion. Insulin loaded into the FL may protect insulin from the enzymatic degradation in the gastrointestinal tract. In addition, the direct introduction of insulin into the epithelial cells by FL may increase the insulin local concentration in the epithelial cells and may pass through the intestinal membrane without detectable cellular damage (i.e., the absence of LDH leakage). Therefore, our results indicated that the direct insulin delivery across the epithelial membrane by FL is essential for improving the absorption of insulin from the intestine. The high bioavailability of insulin represents potential for mucosal delivery carrier.
Conclusion
The present findings suggest that FL enhanced the insulin absorption from the large intestine without causing detectable mucosal damage. The FL's absorption-enhancing effect of insulin mucosal absorption showed site specificity because the mucous/glycocalyx layers on the small intestinal mucosa impeded the accessibility of FL to the intestinal mucosa. The absorption-enhancement effect of insulin was strongly influenced by the loading amount of insulin into FL. Insulin absorption was further increased by the coadministration of IDE inhibitor, indicating that the insulin introduced into the epithelium may be degraded by the IDE present in the cytoplasm. These results imply that FL can potentially be developed as a carrier for improving insulin absorption from the large intestine. The FL may offer a novel mucosal delivery method not only for insulin but also for any kind of macromolecules, such as proteins and gene, as long as they can be encapsulated into the liposomes. Moreover, it is thought that FL is adapted to the large-scale industrial production (24). We believe that this technology may lead to the achievement of an ideal delivery system for macromolecular therapeutic drugs.
References
M. Morishita I. Morishita K. Takayama Y. Machida T. Nagai (1993) ArticleTitleSite-dependent effect of aprotinin, sodium caprate, Na2EDTA and sodium glycocholate on intestinal absorption of insulin Biol. Pharm. Bull. 16 68–72 Occurrence Handle1:CAS:528:DyaK3sXisVyisbo%3D Occurrence Handle7690292
M. A. Radwant H. Y. Aboul-Enein (2002) ArticleTitleThe effect of oral absorption enhancers on the in vivo performance of insulin-loaded poly(ethylcyanoacrylate) nanospheres in diabetic rats J. Microencapsul. 19 225–235 Occurrence Handle1:STN:280:DC%2BD387gsV2rug%3D%3D Occurrence Handle11837977
I. Morishita M. Morishita K. Takayama Y. Machida T. Nagai (1992) ArticleTitleHypoglycemic effect of novel oral microspheres of insulin with protease inhibitor in normal and diabetic rats Int. J. Pharm. 78 9–16 Occurrence Handle1:CAS:528:DyaK38Xks1Wlug%3D%3D
H. Asada T. Douen M. Waki S. Adachi Y. Fujita A. Yamamoto S. Muranishi (1995) ArticleTitleAbsorption characteristics of chemically modified-insulin derivatives with various fatty acids in the small and large intestine J. Pharm. Sci. 84 682–687 Occurrence Handle1:CAS:528:DyaK2MXlslyktLo%3D Occurrence Handle7562404
C. Q. Xia J. Wang W. C. Shen (2000) ArticleTitleHypoglycemic effect of insulin-transferrin conjugate in streptozotocin-induced diabetic rats J. Pharmacol. Exp. Ther. 295 594–600 Occurrence Handle1:CAS:528:DC%2BD3cXnsl2ksbc%3D Occurrence Handle11046093
M. A. Kisel L. N. Kulik I. S. Tsybovsky A. P. Vlasov M. S. Vorob'yov E. A. Kholodova Z. V. Zabarovskaya (2001) ArticleTitleLiposomes with phosphatidylethanol as a carrier for oral delivery of insulin: studies in the rat Int. J. Pharm. 216 105–114 Occurrence Handle10.1016/S0378-5173(01)00579-8 Occurrence Handle1:CAS:528:DC%2BD3MXitFyru7k%3D Occurrence Handle11274812
H. Takeuchi H. Yamamoto T. Niwa T. Hino Y. Kawashima (1996) ArticleTitleEnteral absorption of insulin in rats from mucoadhesive chitosan-coated liposomes Pharm. Res. 13 896–901 Occurrence Handle10.1023/A:1016009313548 Occurrence Handle1:CAS:528:DyaK28Xjs1yrt7s%3D Occurrence Handle8792429
K. Kato M. Nakanishi Y. Kaneda T. Uchida Y. Okada (1991) ArticleTitleExpression of hepatitis B virus surface antigen in adult rat liver. Co-introduction of DNA and nuclear protein by a simplified liposome method J. Biol. Chem. 266 3361–3364 Occurrence Handle1:CAS:528:DyaK3MXhsFynt74%3D Occurrence Handle1847370
A. Hayashi T. Nakanishi J. Kunisawa M. Kondoh S. Imazu Y. Tsutsumi K. Tanaka H. Fujiwara T. Hamaoka T. Mayumi (1999) ArticleTitleA novel vaccine delivery system using immunopotentiating fusogenic liposomes Biochem. Biophys. Res. Commun. 261 824–828 Occurrence Handle10.1006/bbrc.1999.1044 Occurrence Handle1:CAS:528:DyaK1MXltVahu7k%3D Occurrence Handle10441509
J. Kunisawa T. Masuda K. Katayama T. Yoshikawa Y. Tsutsumi M. Akashi T. Mayumi S. Nakagawa (2005) ArticleTitleFusogenic liposome delivers encapsulated nanoparticles for cytosolic controlled gene release J. Control. Release 105 344–353 Occurrence Handle10.1016/j.jconrel.2005.03.020 Occurrence Handle1:CAS:528:DC%2BD2MXlvVyrtbw%3D Occurrence Handle15936842
A. Frey K. T. Giannasca R. Weltzin P. J. Giannasca H. Reggio W. I. Lencer M. R. Neutra (1996) ArticleTitleRole of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting J. Exp. Med. 184 1045– 1059 Occurrence Handle10.1084/jem.184.3.1045 Occurrence Handle1:CAS:528:DyaK28XlsFaltrs%3D Occurrence Handle9064322
G. Wang G. Williams H. Xia M. Hickey J. Shao B. L. Davidson P. B. McCray (2002) ArticleTitleApical barriers to airway epithelial cell gene transfer with amphotropic retroviral vectors Gene Ther. 9 922–931 Occurrence Handle1:CAS:528:DC%2BD38Xksl2mtrs%3D Occurrence Handle12085240
M. Morishita Y. Aoki M. Sakagami T. Nagai K. Takayama (2004) ArticleTitle In situ ileal absorption of insulin in rats: effects of hyaluronidase pretreatment diminishing the mucous/glycocalyx layers Pharm. Res. 21 309–316 Occurrence Handle10.1023/B:PHAM.0000016244.88820.28 Occurrence Handle1:CAS:528:DC%2BD2cXhtlWqtrk%3D Occurrence Handle15032313
Y. Aoki, M. Morishita, K. Asai, B. Akikusa, S. Hosoda, and K. Takayama. Region-dependent role of the mucous/glycocalyx layers in insulin permeation across rat small intestinal membrane. Pharm Res 22:1854–1862 (in press).
J. P. Bai L. L. Chang (1995) ArticleTitleTransepithelial transport of insulin: I. Insulin degradation by insulin-degrading enzyme in small intestinal epithelium Pharm. Res. 12 1171–1175 Occurrence Handle1:CAS:528:DyaK2MXnsVKjs78%3D Occurrence Handle7494830
J. P. Bai L. L. Chang (1996) ArticleTitleEffects of enzyme inhibitors and insulin concentration on transepithelial transport of insulin in rats J. Pharm. Pharmacol. 48 1078–1082 Occurrence Handle1:CAS:528:DyaK2sXhs1GltA%3D%3D Occurrence Handle8953512
J. Hari K. Shii R. A. Roth (1987) ArticleTitle In vivo association of [125I]-insulin with a cytosolic insulin-degrading enzyme: detection bycovalent cross-linking and immunoprecipitation with a monoclonal antibody Endocrinology 120 829–831 Occurrence Handle1:CAS:528:DyaL2sXhtFeqs78%3D Occurrence Handle3026782 Occurrence Handle10.1210/endo-120-2-829
M. Nakanishi T. Uchida H. Sugawa M. Ishiura Y. Okada (1985) ArticleTitleEfficient introduction of contents of liposomes into cells using HVJ (Sendai virus) Exp. Cell Res. 159 399–409 Occurrence Handle10.1016/S0014-4827(85)80013-6 Occurrence Handle1:CAS:528:DyaL28Xhs1WgsQ%3D%3D Occurrence Handle2993007
Y. Okada (1988) ArticleTitleSendai virus-mediated cell fusion Curr. Top. Membr. Transp. 32 297–336
P. Langguth V. Bohner J. Heizmann H. P. Merkle S. Wolffram G. L. Amidon S. Yamashita (1997) ArticleTitleThe challenge of proteolytic enzymes in intestinal peptide delivery J. Control. Release 46 39–57 Occurrence Handle10.1016/S0168-3659(96)01586-6 Occurrence Handle1:CAS:528:DyaK2sXhtlyhsL8%3D
A. M. Ugolev L. F. Smirnova N. N. Iezuitova N. M. Timofeeva N. M. Mityushova V. V. Egorova E. M. Parshkov (1979) ArticleTitleDistribution of some adsorbed and intrinsic enzymes between the mucosal cells of the rat small intestine and the apical glycocalyx separated from them FEBS Lett. 104 35–38 Occurrence Handle10.1016/0014-5793(79)81080-7 Occurrence Handle1:CAS:528:DyaE1MXltlOrtbg%3D Occurrence Handle314388
K. Asano T. Murachi A. Asano (1983) ArticleTitleStructural requirements for hemolytic activity of F-glycoprotein of HVJ (Sendai virus) studied by proteolytic dissection J. Biochem. 93 733–741 Occurrence Handle1:CAS:528:DyaL3sXhslGrurY%3D Occurrence Handle6307987
M. C. Pedroso de Lima J. Ramalho-Santos M. F. Martins A. Pato de Carvalho V. Bairos S. Nir (1992) ArticleTitleKinetic modeling of Sendai virus fusion with PC-12 cells. Effect of pH and temperature on fusion and viral inactivation Eur. J. Biochem. 205 181–186 Occurrence Handle10.1111/j.1432-1033.1992.tb16766.x Occurrence Handle1:STN:280:DyaK383gvFGjsw%3D%3D Occurrence Handle1313363
M. Nakanishi (2005) Biological and chemical hybrid vectors K. Taira K. Kataoka T. Niidome (Eds) Non-Viral Gene Therapy Springer-Verlag Tokyo 118–132
Acknowledgments
We thank Prof. Tomoko Takahashi and Dr. Mayumi Ikegami at the Institute of Medicinal Chemistry, Hoshi University, for their support in the preparation of transverse frozen sections by cryostat.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goto, T., Morishita, M., Nishimura, K. et al. Novel Mucosal Insulin Delivery Systems Based on Fusogenic Liposomes. Pharm Res 23, 384–391 (2006). https://doi.org/10.1007/s11095-005-9175-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-005-9175-7