
Abstract
Matricellular proteins (MCPs) are actively expressed non-structural proteins present in the extracellular matrix, which rapidly turnover and possess regulatory roles, as well as mediate cell–cell interactions. MCPs characteristically contain binding sites for other extracellular proteins, cell surface receptors, growth factors, cytokines and proteases, that provide structural support for surrounding cells. MCPs are present in most organs, including brain, and play a major role in cell–cell interactions and tissue repair. Among the MCPs found in brain include thrombospondin-1/2, secreted protein acidic and rich in cysteine family (SPARC), including Hevin/SC1, Tenascin C and CYR61/Connective Tissue Growth Factor/Nov family of proteins, glypicans, galectins, plasminogen activator inhibitor (PAI-1), autotaxin, fibulin and perisostin. This review summarizes the potential role of MCPs in the pathogenesis of major neurological disorders, including Alzheimer’s disease, amyotrophic lateral sclerosis, ischemia, trauma, hepatic encephalopathy, Down’s syndrome, autism, multiple sclerosis, brain neoplasms, Parkinson’s disease and epilepsy. Potential therapeutic opportunities of MCP’s for these disorders are also considered in this review.
Similar content being viewed by others
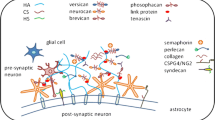

Introduction
The extracellular matrix (ECM) represents a collection of extracellular molecules secreted by cells, that provide structural and biochemical support to the surrounding cells. The ECM includes both the interstitial matrix and the basement membrane. Polysaccharides and fibrous proteins are the main constituents of the interstitial space. The basement membrane is a sheet-like deposition of ECM on which various epithelial cells rest. The ECM is composed of two main classes of macromolecules: proteoglycans and fibrous proteins (e.g., collagens, elastins, fibronectins and laminins) that are required for tissue morphogenesis, differentiation and homeostasis [1]. Many components of the extracellular matrix have been identified in brain (e.g., neural cell adhesion molecules, integrins, laminins, fibronectin, collagens, among others) and all have been implicated in the pathogenesis of several neurological disorders [2–5].
A number of extracellular matrix glycoproteins are expressed in the developing and adult nervous system. Neural stem cells, neurons, and glia express receptors that mediate interactions with specific extracellular matrix molecules. In contrast to other tissues, the ECM of the CNS lacks fibrous proteins under normal conditions. Instead, the neural ECM is rich in glycoproteins and proteoglycans. It has been estimated that the neural ECM makes up approximately 20% of the CNS parenchyma [6]. In vivo genetic and in vitro functional studies have provided evidence that the extracellular matrix critically affects virtually all aspects of nervous system development and function [7].
Matricellular proteins (MCPs) are dynamically expressed non-structural proteins that are present in the ECM [8, 9]. These proteins are rapidly turned-over and possess regulatory roles, rather than serving as stable structural elements of the ECM. They characteristically contain binding sites for ECM structural proteins and cell surface receptors, and are involved in sequestration and modulatory activities of specific growth factors [10]. Examples of matricellular proteins include the CCN family of proteins (also known as CCN intercellular signaling proteins), fibulins, osteopontin, periostin, SPARC family members, tenascin(s), and thrombospondins. Many of these proteins additionally have critical functions in wound healing and tissue repair.
MCPs in brain play a major role in developmental processes, as they are critically involved in the maintenance of neuronal integrity. Changes (increased or decreased) in levels of thrombospondins (TSPs), secreted protein acidic and rich in cysteine family (SPARC), including Hevin/SC1 (SPARC-like 1), Tenascin C (TNC), CCN 2 and 3, glypicans 4 and 6, galectins, plasminogen activator inhibitor-1 (PAI-1), autotaxin, fibulin and perisostin have also been strongly implicated in the pathogenesis of various neurological disorders. SPARC, tenascin, TSP-1 and CCN are known to be upregulated during axonal regeneration, glial scar formation, angiogenesis, and in the “rewiring” of neural circuitry following various neurological conditions. Conversely, decreased levels of MCPs have been strongly implicated the pathogenesis of various neurological conditions, including Alzheimer’s disease, amyotrophic lateral sclerosis, ischemia, traumatic inquiry, hepatic encephalopathy, Down’s syndrome, autism, multiple sclerosis, brain neoplasms, Parkinson’s disease and epilepsy. This review discusses the potential role of MCPs in various neurological disorders, as well as the possibility of targeting MCP’s for promising therapy of these conditions.
Thrombospondin-1/2 (TSP-1/2)
Thrombospondins are a family of multifunctional secreted glycoproteins with antiangiogenic properties [11]. The family consists of thrombospondins 1–5, of which TSP-1 and TSP-2 are produced by astrocytes, that promotes the development of new synapses, as well as maintain neuronal integrity.
TSP-1, also known as THBS1 or THP1 protein, is a member of the thrombospondin family that in humans is encoded by the THBS1 gene [12, 13]. It was first isolated from platelets that had been stimulated with thrombin, and was therefore referred to as the thrombin-sensitive protein, thrombospondin. TSP-1 binds to fibrinogen, fibronectin, laminin, type V collagen, and integrins alpha-V/beta-1, which initiate cell–cell, and cell–matrix interactions [14]. Its major function is the inhibition of the cell proliferation and migration of endothelial cells by interactions with CD36 expressed on the surface of these cells.
While TSP-1 was first recognized for its involvement in angiogenesis, other functions for TSP-1 were subsequently recognized, including apoptosis, activation of TGF-β and immune regulation in peripheral tissues. TSP-1 expression was also identified in the central nervous system (CNS). Post-natal and young adult animal brains, as well as normal human cortical astrocytes, were shown to express TSP-1 [15–19] (see Fig. 1). Cultured astrocytes were also shown to synthesize and secrete TSP-1 [16, 18–21]. Under normal conditions, TSP-1 causes an increase in the total number of synapses [20, 22, 23]. Astrocyte-derived TSP-1 was also shown to mediate the development of pre-synaptic plasticity in vitro [24]. Conversely, defective astrocytic TSP-1 release is known to be associated with neuronal dysfunction (see “Role of TSP-1/2 in Neurological Disorders”).

This figure illustrates the presence of TSP-1 in normal adult rat brain cortical astrocytes ([16], pp 333–347, and cover page). Glial fibrillary acidic protein (GFAP, red, astrocytes), TSP-1 (green), and DAPI (blue, nuclei). The co-localization (merged) image of TSP-1 and GFAP illustrates the enrichment of TSP-1 in astrocytes. Scale bar 20 µm
TSP-2, also known as THBS2, is a disulfide-linked homotrimeric glycoprotein derived from a member of the thrombospondin family that mediates cell–cell, and cell-matrix interactions, and which has a high structural similarity to TSP-1. Similar to TSP-1, TSP-2 binds to extracellular matrix ligands, including transforming growth factor-beta-1, histidine-rich glycoprotein, TSG6, heparin, matrix metalloproteinase-2, and heparan sulfate proteoglycans [25]. TSP-2 also binds to cell surface receptors, including CD36, CD47, LDL receptor-related protein-1 (via calreticulin) and the integrins alpha-V/beta-3, alpha-4/beta-1, and alpha-6/beta-1 [26, 27]. Such binding results in the regulation of proliferation, adhesion, and migration of a number of normal and transformed cell types [28]. TSP-2 is characterized by a distinct regulation of gene expression by various growth factors and hormones. Similar to TSP-1, TSP-2 is highly expressed in developing blood vessels, suggesting a potential role in the regulation of angiogenesis [29, 30]. In contrast to TSP1, TSP2 does not activate latent TGFβ-1, but similar to TSP1 [31], TSP2 contains EGF-like segments that bind calcium in a cooperative manner [32, 33]. Targeted disruption of the TSP-2 gene in mice was shown to increase the number of small- and medium-sized blood vessels in various tissues [34]. TSP-2 expression has been identified in the CNS, particularly in astrocytes under normal conditions [20, 22, 35], and shares a similar sequence homology with TSP-1.
Role of TSP-1/2 in Neurological Disorders
Alzheimer’s Disease (AD)
The level of TSP-1 expression was shown to be decreased in brains of patients with AD [36]. Following treatment of cultured human astrocytoma cells (U373MG), as well as primary cultures of rat brain astrocytes with amyloid beta (Aβ), levels of secreted astrocytic TSP-1 were found to be decreased [18, 36]. In addition, treatment of cultured neurons with Aβ was shown to induce synaptic pathology, including decreased dendritic spine density and reduced synaptic activity [36]. These events were prevented by co-incubation of Aβ with recombinant TSP-1, the latter acting through the alpha-2-delta-1 TSP-1 receptor present on neurons [36]. Further, injection of TSP-1 into the subicula of the hippocampus of an AD model in mice, mitigated the Aβ-mediated reduction of synaptic proteins and related signaling pathways, suggesting that TSP-1 may be a useful therapeutic agent for AD [36]. Additionally, Rama Rao et al. [18] showed that exposure of astrocyte cultures to Aβ1-42 caused a significant (1-3-fold) increase in intracellular levels of TSP-1 that was associated with a reduction in its release. These authors reported that the addition of conditioned media derived from Aβ-treated cultured astrocytes, containing reduced levels of TSP-1 to cultured neurons, resulted in a significant loss of synaptophysin and the postsynaptic density protein 95 (PSD95). These findings suggest that an Aβ-mediated reduction in astrocytic TSP-1 release, contributes to the loss of synaptic integrity in AD, and that strategies aimed at enhancing the astrocytic release of TSP-1 may have therapeutic efficacy in AD.
While TSP-2 is also present in astrocytes and shares a similar sequence homology with TSP-1, its levels vary in different neurological conditions (e.g., decreased TSP-1 release was identified in experimental models of AD, whereas increased TSP-2 expression was detected in brains of patients with AD) [37].
Hepatic Encephalopathy (HE)
HE is the neurological disorder associated with severe liver disease, which presents in acute and chronic forms. Acute HE (AHE) occurs following massive liver necrosis due to viral hepatitis, acetaminophen toxicity, or exposure to various hepatotoxins [38]. It presents with severe brain edema, increase in intracranial pressure, and brain herniation resulting in a high mortality rate [39]. On the other hand, chronic HE (CHE), is usually the consequence of cirrhosis of the liver, generally associated with alcoholism, and presents predominantly with neuropsychiatric symptoms [40]. Patients with CHE exhibit a wide range of neurological symptoms, including altered mood, increased irritability, alterations in sleep/wake cycles, changes in muscle tone and cognitive impairments [40]. A major etiological factor in CHE is the presence of elevated blood and brain levels of ammonia, due to the inability of the diseased liver to significantly detoxify this substance [41].
The principal histopathological finding in CHE is the presence of an astrocytic abnormality referred to as Alzheimer type II astrocytes [42, 43]. The means by which astrocyte dysfunction contributes to the neurological abnormalities in CHE is poorly undertsood. As astrocytes are involved in synapse formation during development, as well as in their maintenance in adults [44], a dysfunction in these cells has been postulated to significantly contribute to the neurobehavioral abnormalities associated with CHE [45, 46]. It has been recently reported that the synaptic dysfunction induced by defective astrocytes may largely occur through a reduction in their levels of MCPs [16, 47].
It was previously shown that chronic ammonia exposure in cultured astrocytes results in decreased intra- and extracellular levels of TSP-1 [16]. A reduction in synaptophysin and PSD95 levels where identified in cultured neurons after exposure to conditioned media (CM) derived from ammonia-treated astrocytes, and such reduction was diminished when recombinant TSP-1 (rTSP-1) was added to the CM of ammonia-treated astrocytes [16]. Further, increased c-Myc content (which is known to inhibit TSP-1 synthesis) was identified in ammonia-treated cultured astrocytes, while silencing c-Myc or pharmacologically inhibiting c-Myc significantly enhanced intra- and extracellular levels of TSP-1 after ammonia treatment of these cultures [16]. Additionally, exposure of cultured neurons to CM from ammonia-treated astrocytes, in which c-Myc was silenced or inhibited by 10058-F4, caused a reversal of the synaptophysin loss. These findings suggest that an ammonia-induced reduction in astrocytic TSP-1 results in a decline in synaptic protein levels, which ultimately contributes to the pathogenesis of CHE [16].
Ischemia
Lin et al. [48] first demonstrated the presence of TSP-1 follwing the middle cerebral artery occlusion (MCA) model of ischemia in rats. These authors identified a biphasic expression of TSP-1 after ischemia (peaking at 1 and 72 h), particularly in meningeal endothelial cells, whereas glial, neuronal, and macrophage cells only expressed TSP-1 at 6 weeks after ischemia. In a similar model of ischemia in mice, Hayashi et al. [49], however, found that TSP-1 level increased within one hour after MCA occlusion, whereas its level decreased from 24 h to 21 days. While these authors used an identical animal model of ischemia, the reason for the differences in TSP-1 expression in these studies is unclear. Further, oxygen-glucose deprivation of murine cerebral endothelial cells in vitro was shown to increase the extent of methylation of the promoter region of TSP-1 with a concomitant decrease in TSP-1 mRNA and protein expression [50]. Similar findings were also reported by Hayashi et al. [49].
Following permanent MCA occlusion in mice, TSP-1 levels were shown to be increased in astrocytes [51]. Further, TSP-1 knock-out (KO) mice displayed a significant deficit in their ability to recover motor function as compared to wild-type (WT) mice [51]. Moreover, TSP-1 KO mice exhibited significant deficits in synaptic density and axonal sprouting, suggesting that a deficiency of TSP-1 results in impaired recovery after stroke [51]. Additionally, plasma TSP-1 level was shown to be increased in the cerebral ischemia associated with sickle cell anaemia [52]. Increased TSP-1 levels were also identified in copper-induced cerebral ischemic injury in mice [53], and in the plasma of patients with ischemic stroke [54], suggesting a potential role of TSP-1 in diminishing the extent of ischemic brain injury.
While the regulatory mechanism of TSP-1 in brain has not been fully investigated, Cekanaviciute et al. [55] reported that the expression of TSP-1 is tightly controlled by TGF-β as “Ast-Tbr2DN” mice, in which TGFβ signaling was specifically inhibited in astrocytes, were shown to inhibit TSP-1 expression, following middle cerebral artery occlusion. Additionally, Xing et al. [56] reported that lipocalin-2 enhances TSP-1 levels in cultured astrocytes.
Although TSP-1 expression was identified in almost all neural cells following ischemia, TSP-2 mRNA and protein levels were observed in endothelial cells, neurons, and macrophages, but not in glial cells [48]. Noteworthy, increasedTSP-1 expression was found in the early stage of ischemia (1–24 h), while TSP-2 overexpression occurred only several weeks after ischemia. While the role of TSP-1 has been extensively examined in ischemia, the involvement of TSP-2 in ischemia remains poorly understood.
Traumatic Brain Injury (TBI)
Increased levels of TSP-1 were observed in rat brain following TBI [57], as well as in cultured astrocytes after a scratch-induced injury [21]. We recently reported that exposure of cultured rat astrocytes to trauma (fluid-percussion injury) increased the levels of intra- and extracellular TSP-1 at early stages, whereas a reduction in such levels was observed at later stages. Additionally, a reduction in NMDA-nr1 and PSD95 levels were identified in cultured neurons post-trauma, while exposure of these traumatized neurons to CM from traumatized astrocytes derived from early stages of trauma, blocked the trauma-induced reduction in NMDA-nr1 and PSD95 levels. These findings suggest that TSP-1 prevents the loss of neuronal integrity following TBI.
Down’s Syndrome (DS) and Autism
Using cultured human astrocytes derived from DS fetal tissue, as well as cultured rat hippocampal astrocytes, Garcia et al. [58] reported that DS astrocytes are directly involved in the development of spine malformations and in a reduction in synaptic density. The authors further documented a deficit in astrocytic TSP-1 in brains from patients with DS, and that such depletion of TSP-1 resulted in abnormalities in spine morphology, whereas the addition of recombinant TSP-1 to neurons prevented such abnormalities. Furthermore, the addition of media from astrocyte cultures derived from TSP-1 KO mice to cultured neurons exhibited similar deficits in spine formation and structure [58].
Autism is a severe neurodevelopmental disorder associated with impaired social interactions and language communication. The mechanisms involved in the pathogenesis of autism are still unclear. Since the TSP-1 gene has been shown to play a critical role in synaptogenesis in the developing brain, Lu et al. [59] investigated the role of the TSP-1 gene in autism by analyzing the coding exons and the 5′-untranslated region (UTR) of the TSP-1 gene. The authors found that both common and rare variants of the TSP-1 gene were indeed associated with autism.
Parkinson’s Disease
Astrocytes derived from embryonic glial-restricted precursor cells by exposure to bone morphogenetic protein (GDAsBMP), which are known to synthesize and release greater levels of TSP-1 than wild-type astrocytes, when transplanted into the 6-hydroxydopamine lesioned rat striatum restored tyrosine hydroxylase expression, as well as promoted behavioral recovery [60]. A similar transplantation also reversed pathological changes, including the loss of parvalbumin-positive GABAergic interneurons, likely by the increased synthesis and release of TSP-1 [60].
Neoplasms
TSP-1 secretion was shown to be reduced in neoplasms derived from breast, bladder, fibroblasts and other organs, resulting in the induction of neovascularization required for tumor growth and metastasis. Full-length and short TSP-1-derived peptides were shown to inhibit angiogenesis by inducing endothelial cell apoptosis and thereby disrupt the vasculature of the growing tumor [61]. However, studies have shown that malignant glioma cells secrete high levels of TSP-1, which may be involved in the migration of glioma cells via interactions with alphavbeta3 and alpha3beta1 integrins [62], and that treatment of rats bearing either C6 or 9L tumors with TSP1-derived peptide D-reverse amKRFKQDGGWSHWSPWSSac was shown to retard brain tumor growth, likely as a result of slower de novo blood vessel formation and synergistic antiproliferative effects on tumor cells [63]. Further, loss of tumor suppressors on chromosome 10 was shown to contribute to the aggressive malignancy of glioblastomas, in part by the inhibition of angiogenesis by TSP-1 in lower grade tumors [64]. TSP-1 over-expression was slso shown to occur in a differentiation-specific manner in neuroblastic tumors [65]. Lastly, mutation of the p53 gene provides gliomas with an angiogenic phenotype by reducing thrombospondin-1 production, as well as by enhancing angiogenesis inducers in the early phase of malignant progression [66].
Other Conditions
TSP-1 expression was identified before the peak of angiogenesis, whereas a robust TSP-2 expression occurred at the peak of angiogenesis which continued until angiogenesis was completed. Using purified rat retinal ganglion cells (RGCs), Christopherson et al. [20] found that TSP-2 mimicked the ability of astrocyte conditioned media to increase the number of excitatory synapses in cultured RGCs. Additionally, immunodepletion of TSP-2 from astrocyte conditioned media prevented the astrocyte-induced synaptogenesis, suggesting that TSP-2 is a necessary astrocytic signal for the generation of excitatory synapses in RGCs. Collectively, these findings suggest that astrocytic TSP-1/2 are necessary signals involved in the maintenance of neuronal integrity.
Therapeutic Implications
Since TSP-1 is a large protein (~120 kDa), its potential clinical use is limited. However, agents that enhance or decrease their level in astrocytes have recently been targeted for the treatment of hepatic encephalopathy, cerebral ischemia, TBI, AD and neoplasms [16, 36, 51, 57, 67]. Additionally, small-molecules based on a CD36-binding peptide sequence derived from TSP1 are currently being tested for the treatment of various neurological disorders, including ischemia and neoplasms [67, 68]. One analog of TSP-1, ABT-510, exhibited potent pro-apoptotic activity in glioma cell lines [67], and was subsequently shown to display therapeutic benefit in several CNS malignancies [67].
Tenascin-C
Tenascin C (TNC) is a large hexameric extracellular glycoprotein that is expressed in the extracellular matrix. TNC is classified as an adhesion-modulating protein, as it was found to inhibit the cellular adhesion to fibronectin [69]. TNC is expressed in restricted perivascular areas of the CNS during development, disease or injury. In the developing CNS, TNC is first expressed by radial glia and later primarily by astrocytes, where it appears to exert autocrine effects involved in the regulation of astrocyte progenitor cell proliferation [70]. Its expression has also been identified in postnatal stages, as well as in the adult CNS [71, 72]. TNC contributes to the maturation of neural progenitor cells and to the proliferation and maintenance of oligodendrocyte precursors [73–75]. The TNC-regulated target gene, Sam68, is involved in the control of glial cell proliferation [76]. In vivo and in vitro studies further shows that TNC encodes latent, as well as inhibitory signals that mediate neuron migration and axon growth and guidance in the context of neuron-glia interactions [77–83]. TNC has been implicated in inflammation and tumor development in the CNS [84, 85] (see below for its expression status and specific functions in various neurological conditions).
Role in Neurological Disorders
Traumatic Brain Injury
Using in situ hybridization and immunocytochemistry, Laywell et al. [86] showed that stab wounds of the adult mouse cerebellar and cerebral cortices result in enhanced expression of TNC around the lesion site which was associated with a subset of astrocytes. The authors speculated that TNC up-regulation in the lesioned adult brain may be directly involved in failed regeneration, or indirectly involved through interactions with other glycoconjugates that either inhibit or facilitate neurite outgrowth. Hausmann and Betz [87] investigated the time-dependent vascular response during the first 30 weeks after TBI in humans and found a significantly increased astrocytic TNC immunoreactivity at 1.6 days following injury.
In a scratch model of trauma in cultured astrocytes, Nishio et al. and Wanner et al. [88, 89] demonstrated increased synthesis and release of TNC, as well as increased expression of TNC mRNA. Further, Nishio et al. [88] showed that the exogenous application of TNC to cultured astrocytes enhanced cell proliferation and migration, while its functional blocking with anti-TNC or anti-integrin beta-1 antibodies reduced such events. These findings suggest that mechanical injury induces the astrocytic production and release of TNC leading to astrocyte proliferation and migration. Increased TNC level has also been identified following cortical penetrating stab wounds in mice [90].
While astrocytes were shown to highly express TNC, Yu et al. [91] documented the upregulation of TNC in regenerating neurons of the nucleus of the medial longitudinal fasciculus (NMLF) in brainstem and spinal motoneurons. Further, inhibition of TNC expression by an antisense oligonucleotide resulted in impaired locomotor recovery, reduced regrowth of axons and diminished synapse formation by the regrowing spinal motoneurons, all vital indicators of regeneration. These findings suggest a promising role for TNC in promoting spinal cord regeneration following injury. For a comprehensive review on the role of TNC following injury to CNS [92].
Although the precise role of TNC in response to injury remains unclear, Zhang et al. [93] and Deller et al. [94] reported that regenerating spinal cord and entorhinal cortical neurons were closely associated with TNC expression, suggesting that TNC may be involved in axonal regrowth. Additionally, Ikeshima-Kataoka et al. [95] showed that in tenascin-deficient mice, the expression of interleukin (IL)-1β, tumor necrosis factor-α and IL-6 were higher, whereas levels of IL-4 and granulocyte colony-stimulating factor were lower in wild-type mice, suggesting that TNC also aids in the production of inflammatory cytokines in the injured brain. However, Gates et al. [96] reported that TNC inhibits neurite regeneration following injury, and that blocking TNC with antibodies enhanced neurite growth, suggesting that TNC release from astrocytes may detrimental for neurite outgrowth.
Alzheimer’s Disease (AD)
Increased plasma levels of TNC [97], as well as TNC plaques in reactive astrocytes and microglia along with phosphorylated tau-containing dystrophic neurites of amyloid cores were observed in patients with AD, suggest a role for TNC in Aβ plaque formation [98]. Further, Xie et al. [99] observed that TNC gene transcription was upregulated in cultured microglia after Aβ challenge, as well as in brains of transgenic mice (Tg) overexpressing mutated amyloid precursor protein (APP) in neural cells. Additionally, these authors showed that TNC deficiency induced by cross-breeding APP-Tg mice and TNC-deficient mice, reduced pro-inflammatory events and enhanced anti-inflammatory activation in the mutated APP-Tg mice brain. These changes were associated with a reduced cerebral Aβ content and higher levels of the PSD95. These findings suggest that a “functional” inhibition of TNC may exert beneficial effects in AD.
Multiple Sclerosis (MS)
Gutowski et al. [100] reported a major loss of TNC at the edge of acute MS plaques. In subacute lesions, however, TNC-immunopositive reactive astrocytes were prominent, reflecting the synthesis of TNC in subacute lesions. Increased TNC level was also observed in the sera of patients with MS [101]. TNC was also expressed in chronic MS plaques at levels similar to those seen in adjacent white matter. The authors suggested that the loss of TNC in acute plaques was indicative of an enzyme-mediated breakdown of the extracellular matrix, likely secondary to a disruption of the blood–brain barrier and associated leukocyte extravasation. The subsequent production of TNC may result in glial scar formation and thereby obstruct remyelination and axonal repair. It is also, however, possible that reduced astrogliosis resulting from reduced TNC levels in MS may be beneficial for remyelination due to the lesser degree of scar formation [102].
Zendedel et al. [103] examined the means by which astrocytes promote remyelination in chronically demyelinated lesions using the cuprizone mouse model of MS. Treatment of mice with triiodothyronin (which is thought to enhance remyelination by the induction of oligodendrocyte maturation) that had been fed with cuprizone, reduced the astrogliosis, possibly by diminishing TNC levels. These findings suggest that triiodothyronin promotes remyelination in chronic demyelinative lesions by enhancing oligodendrocyte maturation and by attenuating astrogliosis, likely as a consequence of reduced levels of TNC.
Epilepsy
An increase in TNC immunoreactivity (IR) was observed in the hippocampus of epileptic rats that displayed neuronal cell loss and associated gliosis [104]. TNC levels were increased both in reactive astrocytes and in axonal plasma membranes of the hippocampus in these rats. However, TNC-IR in the molecular layer of the hippocampus remained unchanged in kindled or kainate-treated rats. Additionally, the authors observed increased TNC-IR in zones in which axonal regeneration did not occur (the hippocampal CA3 region), whereas zones in which reactive synaptogenesis occurred (the molecular layer of kindled or kainate-treated rats) were devoid of TNC-IR. These findings suggest that TNC may obstruct the terminal sprouting of mossy fibers associated with neuronal loss and gliosis in the hippocampal CA3 region of kainate-treated rats, and that such loss may contribute to epileptogenesis.
TNC expression was also shown to be upregulated in rat hippocampal neurons in the early stages (4–5 h) following kainic acid (KA) injection, which was associated with the activation of granule cells and the sprouting of axon terminals [105]. TNC overexpression was also detected at later stages following KA injection in pyramidal cells in the CA1 and CA3 regions, and such overexpression was associated with reactive gliosis [105]. It is also possible, however, that the stimulation of TNC expression in KA-treated rats may be due to the induction of seizures through the activation of basic fibroblast growth factor [106–109]. A strong upregulation of TNC was also identified in the dentate gyrus, especially in the sprouting molecular layer of a murine model of temporal lobe epilepsy [110], as well as in seizures following pilocarpine administration [111].
In addition to TNC, tenascin-R (TNR) is also expressed in the CNS and has also been implicated in the pathogenesis of epilepsy. Mice deficient in TNR showed increased hippocampal excitability without displaying an increase in susceptibility to seizures in the pilocarpine model of epilepsy [112, 113]. Further, Hoffmann et al. [114] reported that kindling progression was retarded in TNR-/- mice, in which an increase in the number of S100-β expressing astrocytes in the dentate gyrus was enhanced by the deficiency in TNR. These findings suggest that in addition to TNC, TNR may also play a role in epileptogenesis.
Neoplasms
The involvement of tenascin in tumor suppression was first reported by Lee et al. [115, 116], who showed that nude mice given injections of Mab 81C6, an IgG2b immunoglobulin (which defines an epitope of the glioma-associated tenascin) displayed a significant delay in tumor growth. Subsequently, Higuchi et al. [117] reported that the degree of histological malignancy and the level of cell dedifferentiation of human gliomas correlated well with the expression of TNC. The authors suggested that that measurement of TNC in the CSF might be useful in the diagnosis of brain tumors, as well as in the monitoring tumor growth [118]. Additionally, a correlation between tenascin expression and the extent of tumor growth have been reported [119–124]. For references on the distribution of TNC in brain tumors, see [125–133].
Recent studies have established that both increased and decreased levels of TNC expression are associated with higher-grade gliomas [134], but not with low-grade gliomas [135], suggesting that TNC may not be suitable for the diagnosis/treatment of low-grade gliomas [134]. TNC loss-of-function promoted glioblastoma multiforme (GBM) neurosphere cell adhesion and actin cytoskeleton organization without any effect on cell growth in vitro, suggesting that tumor cells with decreased TNC expression may be sensitive to anti-proliferative treatment [136]. TNC also acts as a promoter of invasiveness of brain tumor-initiating cells (stem cells) through the ADAM-9 signaling pathway [122]. These findings suggest that altered levels of TNC in GBM may be a useful prognostic marker for this neoplasm [121].
Based on the above observations, various studies have used an anti-tenascin antibody to prevent malignant glioma growth [137–142]. Some studies instead have shown promising results using an anti-tenascin antibody [143–151]. However, the use of an anti-tenascin antibody for the treatment of gliomas was only rarely effective as relapses occurred within months after treatment [131, 143, 145, 152].
SPARC
The matricellular protein, secreted protein acidic and rich in cysteine (SPARC), also known as osteonectin and BM-40, is a calcium and collagen-binding glycoprotein. The SPARC family consists of four groups, (i) SPARC and hevin; (ii) SMOC 1 and 2; (iii) testicans 1–3; and (iv) and follistatin-like protein (Fstl-1). SPARC is predominantly expressed in astrocytes and microglia of the cerebellum [153], retinal Müller cells, and cerebellar Bergmann glia.
SPARC is expressed in the adult central nervous system. In the cerebellum, SPARC is present in Bergmann glia, with a strong expression along their radial fibres [154]. In the hippocampus, SPARC was observed in astrocytes [154]. The expression of SPARC in a subset of astrocytes, particularly in synaptically enriched areas, suggests a role for SPARC in the maintenance of extracellular matrix integrity in the adult brain [154–156].
SPARC regulates growth factor signaling, extracellular matrix assembly, cell-matrix interactions, as well as cell adhesion, migration, and cell proliferation [157, 158]. SPARC modulates other growth factors, including transforming growth factor-beta (TGF-β), extracellular matrix proteins, including collagen Types I–V, VIII and vitronectin [159]. SPARC has also been shown to regulate integrin-mediated adhesion [159]. The upregulation of SPARC has been associated with ischemia, neoplasms, and other neurological conditions (see below).
Chronic morphine administration frequently leads to altered behavioral responses (referred to as sensitization), which are thought to result from neuronal hyperactivity and accompanying changes to neuronal activity-dependent gene expression [160]. In particular, the repeated administration of morphine to mice was shown to increase locomotor activity, and that morphine-induced increase in SPARC levels in the basolateral amygdala contributed to the establishment of locomotor sensitization [160]. Upregulation of SPARC was also shown to inhibit DNA synthesis in human microvascular endothelial cells that were stimulated by vascular endothelial growth factor (VEGF) [161], suggesting that SPARC modulates the mitogenic activity of VEGF, that may decrease the association of VEGF with its cell-surface receptors.
Alzheimer’s Disease and Ischemic Conditions
Using mass spectroscopy, SPARC-like protein was identified in samples of cerebrospinal fluid from patients with AD, as compared to age-matched controls [162, 163]. Following a focal, noninvasive photothrombotic lesion (a model of ischemic stroke), reactive microglia rapidly downregulate SPARC expression at the lesion site, concomitant with reactive, perilesional astrocytes upregulating SPARC [164]. Additionally, induction of ischemic injury in SPARC null-mice showed enhanced microgliosis in and around the lesion site of the cerebral cortex, which was accompanied by a significantly enhanced functional recovery after lesion. Microglial cultures from adult SPARC null-mice were also shown to proliferate at a greater rate, which was reversed by the addition of SPARC, suggesting that SPARC is an important regulator of microglial proliferation [164]. Further, Baumann et al. [165] reported that transient global brain ischemia resulted in a significant reduction in SPARC levels in brain blood vessels, and that post-ischemic hypothermia stabilized brain vessels, as well as reduced BBB disruption, in part, by preventing the proteolytic degradation of SPARC.
Following a localized injury to the adult rat forebrain, a biphasic induction of a SPARC-related protein mRNA was detected in cortical neurons bordering the lesion site, followed by a more prolonged induction of SPARC in astrocytes proximal to the lesion site [166]. A similar SPARC-related protein induction pattern was observed in the hippocampus in response to a surgical lesion [166].
Neoplasms
SPARC was recently recognized as a prominent marker of gliomas [167]. SPARC is also known to delay tumor growth [168] by the activation of PTEN-mediated Ser78 HSP27 phosphorylation, as well as by the stimulation of the ERK/AKT and FAK/ILK signaling pathways [169–173], or by reduced VEGF secretion [174]. Further, RNA interference against SPARC was shown to promote the growth of human malignant glioma cells [175]. Some reports also indicate that SPARC promotes glioma invasion [168, 176, 177]. The precise role of SPARC in neoplastic progression, however, remains to be better defined.
Other Neurological Conditions
Increased SPARC in the hippocampus was shown to be associated with depression-related behavior [178]. SPARC secretion was also found to control excitatory CNS synaptogenesis [178], as well as glutamate receptor levels at developing synapses through beta-integrin interactions [179]. Additionally, SPARC was shown to stimulate Schwann cells, resulting in the promotion of dorsal root ganglion outgrowth in vitro and in vivo [180]. Retinal glial conditioned media, containing high levels of SPARC, was shown to significantly enhance neurite growth and branching of adult rat dorsal root ganglion (DRG) neurons in culture. Additionally, the transplantation of retinal glia significantly enhanced regeneration of DRG axons after a crush injury in adult rats through a process mediated by SPARC [181].
Glypicans
Glypicans (Gpc’s) belong to the family of heparin sulphate binding glycoproteins, which includes glypicans 1–6. Gpc’s are critically involved in developmental morphogenesis, and have been implicated as regulators of cell-signaling pathways, including Wnt and Hedgehog, fibroblast growth factors and bone morphogenic proteins [182, 183]. Like other MCPs, they are mostly expressed in brain during development. However, Gpc4 and 6 are expressed predominantly in adult brain astrocytes, with Gpc4 expression elevated in the hippocampus and Gpc6 enriched in the cerebellum.
Glypicans were first cloned, isolated and characterized by David et al. [184], Karthikeyan et al. [185]. It was subsequently reported that glypicans are predominantly neuronal membrane components that are present in late embryonic and postnatal CNS [186, 187]. Additionally, Veugelers et al. [188] reported Gpc-5 enrichment in adult human brain tissue, while Saunders et al. [189] showed that Gpc-5 was highly expressed in brain at developmental stages [190]. Glypican mRNA levels were specifically identified in cerebellar granule cells, large motor neurons in the brainstem, and in CA3 pyramidal cells of the hippocampus [187].
There is increasing evidence that Gpc’s act as regulators of cell migration, axonal pathfinding, synaptogenesis, and structural plasticity. In brain, neuronal Gpc are regulated by N-methyl-D-aspartate and metabotropic receptor activation [191]. Gpc’s serve as cell-surface receptors for neurocan, which is involved in the promotion of neurite outgrowth [192]; axon guidance, and visual system function [193]; brain patterning and neurogenesis [194]; cell cycle progression [195], as well as in the regulation of brain size [196].
Alzheimer’s Disease
A role for Gpc’s in AD is well established. Glypican-1 (Gpc-1), was shown to be abundantly expressed in cerebral amyloid angiopathy [197–199]. Gpc-1 was also expressed in neuritic plaques and neurofibrillary tangles in AD [200]. Further, Gpc-1 binds to APP and such binding inhibits APP-induced neurite outgrowth in chick sympathetic neurons [201].
Cultured cortical neurons from APP KO mice showed an increased nitric oxide-mediated degradation of Gpc-1, as compared to brain tissue and neurons from wild-type mice, suggesting a nitroxyl anion-catalyzed degradation of Gpc-1 in AD [202]. Increased levels of Gpc-1 were also identified in astrocyte cultures following exposure to Aβ [203]. Further, when human brain pericytes were cultured in the presence of Aβ, Gpc-1 protein and mRNA expression were increased, suggesting that Aβ modulates the cellular expression of Gpc-1 [204]. Such modulation may prevent Aβ-mediated cellular toxicity in AD, possibly by enhancing heparan sulfate accumulation [204], suggesting that enhanced levels of Gpc’s may be neuroprotective in AD.
Neoplasms
Gpc’s have also been implicated in tumor progression. Gpc-1 is highly expressed in gliomas and such expression was shown to induce DNA replication, suggesting a mutagenic activity for Gpc-1. Gpc-1 expression also led to a significant downregulation of the tumor suppressors pRb, Cip/Kip cyclin-dependent kinase inhibitors (CKIs), and CDH1, as well as to an upregulation of the pro-oncogenic proteins cyclin E, cyclin-dependent kinase 2, Skp2, and Cdt1. These Gpc-1-induced changes were accompanied by a significant reduction in all types of D cyclins [205, 206]. These findings suggest that Gpc-1 plays an important role in glioma [205, 206].
Parkinson’s Disease and Ischemia Conditions
The direct infusion of glypican into an infarct cavity for 7 days, beginning 7 days after middle cerebral artery occlusion in rats, increased levels of the microtubule-associated protein-2 immunoreactivity in the peri-infarct region and ameliorated the middle cerebral artery occlusion associated behavioral abnormalities [207]. Intrastriatal transplantation of Gpc4 into a 6-OHDA rat model of Parkinson’s disease (PD) (which exhibit a reduction in Gpc4 level), was shown to improve motor behavior [208].
Hepatic Encephalopathy
In recent studies, we observed a decrease in Gpc-4 and 6 in ammonia-treated cultured astrocytes (unpublished observations), and in brains of rats treated with thioacetamide (a model of heaptic encephalopathy) by immunofluorescence as described previously by us [16] (Figs. 2, 3). Further, enhancing Gpc-4 and 6 levels by blocking oxidative stress and NF-κB activation in ammonia-treated astrocytes, and adding the CM from such procedure to neurons, diminished the loss of synaptophysin induced by CM from ammonia-treated astrocytes (unpublished observation). Astrocytic Gpc-4 and -6 were also shown to promote the formation of excitatory synapses through the activation of GluA1 AMPA receptors subunit [209–211]. These findings collectively suggest a potential role of Gpc’s in the pathogenesis of various neurological disorders.

Glypican-4 protein levels in astrocytes from cerebral cortex of normal and TAA-treated adult rats. Glial fibrillary acidic protein (GFAP, green, astrocytes), glypican-4 (red), and DAPI (blue, nuclei). The co-localization of glypican-4 and GFAP illustrates the presence of glypican-4 in adult rat brain astrocytes (a–d). Additionally, treatment of rats with the liver toxin TAA, reduced the glypican-4 levels in astrocytes (e–h). I, quantification of glypican-4 levels after TAA. Scale bar 10 µm

Glypican-6 protein levels in astrocytes from cerebral cortex of normal and TAA-treated adult rats. Glial fibrillary acidic protein (GFAP, red, astrocytes), glypican-6 (green), and DAPI (blue, nuclei). The co-localization of glypican-6 and GFAP illustrates the presence of glypican-6 in adult rat brain astrocytes (a–d). Additionally, treatment of rats with the liver toxin TAA, reduced the glypican-6 levels in astrocytes (e–h). I, quantification of glypican-6 levels after TAA. Scale bar 15 µm
CCN Family of Proteins
This family of proteins consists of a complex of six proteins (CCN1-6) encoded from the CCN (CYR61/CTGF/NOV) family of genes. These proteins interact with cell surface receptors and receptor ligands to induce intracellular signaling. They regulate cell adhesion, proliferation, migration, chemotaxis, cell survival, differentiation, angiogenesis, chondrogenesis, tumorigenesis and wound healing [212, 213]. Except CCN6, most of these proteins are present in adult brain. CCN1 is present in the frontal, temporal and occipital cortices, hippocampus and caudate, and is strongly expressed in the spinal cord [214]. CCN2 expression is similar to CCN1, but is lower in the hippocampus, caudate and corpus callosum [215]. It is also present in cortical glial cells [216]. CCN3 is present in cortex, hippocampus, amygdala and spinal cord [214, 217]. CCN4 is found in cerebral cortex, cerebellum, cortical endothelial cells and Purkinje neurons. CCN5 is present in the adult cortex, cerebellum and spinal cord neurons [215 and references therein].
While the presence of CCN family of proteins has been associated with various neurological disorders, including neoplasms, ischemia, amyotrophic lateral sclerosis, trauma and alzheimer’s disease [215], their precise involvement in these conditions is not clear. In addition to the CCN family of proteins, changes in osteopontin, small leucine-rich proteoglycans (SLRPs) in rat anterior pituitary, Fibulin-5 and Galectins 1 and 3 have also been found in various neurological conditions [218–228]. Their role in the pathogenesis in these conditions, however, is not yet well established.
Summary/Perspectives
MCPs play a major role in the pathogenesis of various neurological disorders, including Alzheimer’s disease, amyotrophic lateral sclerosis, ischemia, traumatic brain inquiry, hepatic encephalopathy, Down’s syndrome, autism, multiple sclerosis, neoplasms, Parkinson disease and epilepsy. Increased MCPs levels have also been identified in neoplasms, ischemia and Alzheimer’s disease, while decreased levels were identified in hepatic encephalopathy and traumatic brain inquiry (Table 1). The reversal of MCP levels by genetic manipulation, or by the inhibition of upstream signaling, was shown to reduce disease progression in these conditions.
While the reason for alterations in MCPs expression and their involvement in various neurological disorders remain unclear, it possible that several factors, including the duration and severity of the insult; the concentration of agents used in these investigations to induce a disease process (e.g., Aβ peptides); or the animal model used, may have varied in these studies, possibly accounting for the differences in MCPs protein expression. Regardless, a better understanding of the role of MCPs in these neurological conditions should aid in the identification of novel therapeutic agents for these disorders.
Relative to treatment strategies aimed at diminishing/preventing neurological disorders associated with decreased levels of MCP, agents that enhance such levels have been shown to reduce the severity of these conditions, including, hepatic encephalopathy, traumatic brain injury and cerebral ischemia. Additionally, small-molecules based on a CD36-binding peptide sequence derived from TSP-1 are currently being tested for their efficacy in the treatment of some neurological disorders (e.g., neoplasms, ischemia). One analog of TSP-1, ABT-510, which exhibited potent pro-apoptotic activity in glioma cell lines, was subsequently shown to exhibit therapeutic benefit against several malignancies. ABT-510 is currently in phase II clinical trials for the treatment of various CNS neoplasms (e.g., astrocytomas).
Alterations in MCPs expression appear to be critically involved in the pathogenesis of many neurological conditions, including Alzheimer’s disease, amyotrophic lateral sclerosis, ischemia, trauma, hepatic encephalopathy, Down’s syndrome, autism, multiple sclerosis, neoplasms, Parkinson disease and epilepsy. We anticipate that a better understanding of the role of MCPs in these disorders will provide valuable insights into disease pathogenesis that may ultimately lead to improved therapeutic outcomes for patients with these conditions.
References
Hay ED (1981) Extracellular matrix. J Cell Biol 91(3):205s–223s
Edgar D (1985) Nerve growth factors and molecules of the extracellular matrix in neuronal development. J Cell Sci Suppl 3:107–113
Novak U, Kaye AH (2000) Extracellular matrix and the brain: components and function. J Clin Neurosci 7(4):280–290
Martin GR, Kleinman HK (1985) The extracellular matrix in development and in disease. Semin Liver Dis 5(2):147–156
Sethi MK, Zaia J (2016) Extracellular matrix proteomics in schizophrenia and Alzheimer’s disease. Anal Bioanal Chem. doi:10.1007/s00216-016-9900-6
Nicholson C, Syková E (1998) Extracellular space structure revealed by diffusion analysis. Trends Neurosci 21(5):207–215
Barros CS, Franco SJ, Müller U (2011) Extracellular matrix: functions in the nervous system. Cold Spring Harb Perspect Biol 3(1):a005108
Bornstein P (1995) Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 130(3):503–506
Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14(5):608–616
Roberts DD (2011) Emerging functions of matricellular proteins. Cell Mol Life Sci 68(19):3133–3136
Bornstein P (2009) Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal 3(3–4):189–200
Wolf FW, Eddy RL, Shows TB, Dixit VM (1990) Structure and chromosomal localization of the human thrombospondin gene. Genomics 6(4):685–691
Jaffe E, Bornstein P, Disteche CM (1990) Mapping of the thrombospondin gene to human chromosome 15 and mouse chromosome 2 by in situ hybridization. Genomics 7(1):123–126
Li Z, Calzada MJ, Sipes JM, Cashel JA, Krutzsch HC, Annis DS, Mosher DF, Roberts DD (2002) Interactions of thrombospondins with alpha4beta1 integrin and CD47 differentially modulate T cell behavior. J Cell Biol 157(3):509–519
Asch AS, Leung LL, Shapiro J, Nachman RL (1986) Human brain glial cells synthesize thrombospondin. Proc Natl Acad Sci USA 83(9):2904–2908
Jayakumar AR, Tong XY, Curtis KM, Ruiz-Cordero R, Shamaladevi N, Abuzamel M, Johnstone J, Gaidosh G, Rama Rao KV, Norenberg MD (2004) Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of synaptic proteins: in vitro and in vivo studies. J Neurochem 131(3):333–347
Lu Z, Kipnis J (2010) Thrombospondin 1–a key astrocyte-derived neurogenic factor. FASEB J 24(6):1925–1934
Rama Rao KV, Curtis KM, Johnstone JT, Norenberg MD (2013) Amyloid-β inhibits thrombospondin 1 release from cultured astrocytes: effects on synaptic protein expression. J Neuropathol Exp Neurol 72(8):735–744
Yonezawa T, Hattori S, Inagaki J, Kurosaki M, Takigawa T, Hirohata S, Miyoshi T, Ninomiya Y (2010) Type IV collagen induces expression of thrombospondin-1 that is mediated by integrin alpha1beta1 in astrocytes. Glia 58(7):755–767
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120(3):421–433
Tran MD, Neary JT (2006) Purinergic signaling induces thrombospondin-1 expression in astrocytes. Proc Natl Acad Sci USA 103(24):9321–9326
Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139(2):380–392
Xu J, Xiao N, Xia J (2010) Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci 13(1):22–24
Crawford DC, Jiang X, Taylor A, Mennerick S (2012) Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J Neurosci 32(38):13100–13110
Adams JC (2001) Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol 17:25–51
Petrik JJ, Gentry PA, Feige JJ, LaMarre J (2002) Expression and localization of thrombospondin-1 and -2 and their cell-surface receptor, CD36, during rat follicular development and formation of the corpus luteum. Biol Reprod 67(5):1522–1531
Goicoechea S, Pallero MA, Eggleton P, Michalak M, Murphy-Ullrich JE (2002) The anti-adhesive activity of thrombospondin is mediated by the N-terminal domain of cell surface calreticulin. J Biol Chem 277(40):37219–37228
Lopes N, Gregg D, Vasudevan S, Hassanain H, Goldschmidt-Clermont P, Kovacic H (2003) Thrombospondin 2 regulates cell proliferation induced by Rac1 redox-dependent signaling. Mol Cell Biol 23(15):5401–5408
Lawler PR, Lawler J (2012) Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med 2(5):a006627
Krady MM, Zeng J, Yu J, MacLauchlan S, Skokos EA, Tian W, Bornstein P, Sessa WC, Kyriakides TR (2008) Thrombospondin-2 modulates extracellular matrix remodeling during physiological angiogenesis. Am J Pathol 173(3):879–891
Ribeiro SM, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE (1999) The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J Biol Chem 274(19):13586–13593
Carlson CB, Bernstein DA, Annis DS, Misenheimer TM, Hannah BL, Mosher DF, Keck JL (2005) Structure of the calcium-rich signature domain of human thrombospondin-2. Nat Struct Mol Biol 12(10):910–914
Hoffmann BR, Liu Y, Mosher DF (2012) Modification of EGF-like module 1 of thrombospondin-1, an animal extracellular protein, by O-linked N-acetylglucosamine. PLoS One 7(3):e32762
Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, Iozzo RV, LaMarca M, McKinney CE, Ginns EI, Bornstein P (1998) Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol 140(2):419–430
Scott-Drew S (1997) Expression and function of thrombospondin-1 in myelinating glial cells of the central nervous system. J Neurosci Res 50(2):202–214
Son SM, Nam DW, Cha MY, Kim KH, Byun J, Ryu H, Mook-Jung I (2015) Thrombospondin-1 prevents amyloid beta-mediated synaptic pathology in Alzheimer’s disease. Neurobiol Aging 36(12):3214–3227
Buée L, Hof PR, Roberts DD, Delacourte A, Morrison JH, Fillit HM (1992) Immunohistochemical identification of thrombospondin in normal human brain and in Alzheimer’s disease. Am J Pathol 141:783–788
Norenberg MD, Rama Rao KV, Jayakumar AR (2009) Signaling factors in the mechanism of ammonia neurotoxicity. Metab Brain Dis 24(1):103–117
Jayakumar AR, Rama Rao KV, Norenberg MD (2015) Neuroinflammation in hepatic encephalopathy: mechanistic aspects. J Clin Exp Hepatol 5(Suppl 1):S21–S28
Jones EA, Weissenborn K (1997) Neurology and the liver. J Neurol Neurosurg Psychiatry 63:279–293
Hazell AS, Butterworth RF (1999) Hepatic encephalopathy: An update of pathophysiologic mechanisms. Proc Soc Exp Biol Med 222:99–112
Norenberg MD (1981) The astrocyte in liver disease. In: Fedoroff S, Hertz L (eds) Advances in cellular neurobiology, vol 2. Academic Press, New York, pp 303–352
Martin H, Voss K, Hufnagl P, Wack R, Wassilew G (1987) Morphometric and densitometric investigations of protoplasmic astrocytes and neurons in human hepatic encephalopathy. Exp Pathol 32:241–250
Pfrieger FW (2010) Role of glial cells in the formation and maintenance of synapses. Brain Res Rev 63(1–2):39–46
Gibson G, Zimber A, Krook L, Richardson EJ, Visek W (1974) Brain histology and behaviour of mice injected with urease. J Neuropathol 33:201–211
Norenberg MD (1998) Astroglial dysfunction in hepatic encephalopathy. Metab Brain Dis 13(4):319–335
Albrecht J, Zielińska M (2014) Deficit of astroglia-derived thrombospondin-1 and loss of synaptic proteins in hepatic encephalopathy: do ammonia-overexposed astrocytes derange the synaptic hardware? J Neurochem 131(3):265–277
Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY (2003) Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke 34(1):177–186
Hayashi T, Noshita N, Sugawara T, Chan PH (2003) Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab 23(2):166–180
Hu CJ, Chen SD, Yang DI, Lin TN, Chen CM, Huang TH, Hsu CY (2006) Promoter region methylation and reduced expression of thrombospondin-1 after oxygen-glucose deprivation in murine cerebral endothelial cells. J Cereb Blood Flow Metab 26(12):1519–1526
Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK (2008) Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab 28(10):1722–1732
Faulcon LM, Fu Z, Dulloor P, Barron-Casella E, Savage W, Jennings JM, Van Eyk JE, Debaun M, Casella JF, Everett A (2013) Thrombospondin-1 and L-selectin are associated with silent cerebral infarct in children with sickle cell anaemia. Br J Haematol 162(3):421–424
Jiang Y, Wang LP, Dong XH, Cai J, Jiang GJ, Zhang C, Xie HH (2015) Trace amounts of copper in drinking water aggravate cerebral ischemic injury via impairing endothelial progenitor cells in mice. CNS Neurosci Ther 21(8):677–680
Gao JB, Tang WD, Wang HX, Xu Y (2015) Predictive value of thrombospondin-1 for outcomes in patients with acute ischemic stroke. Clin Chim Acta 450:176–180
Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS (2014) Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 62(8):1227–1240
Xing C, Wang X, Cheng C, Montaner J, Mandeville E, Leung W, van Leyen K, Lok J, Wang X, Lo EH (2014) Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke 45(7):2085–2092
Tran MD, Furones-Alonso O, Sanchez-Molano J, Bramlett HM (2012) Trauma-induced expression of astrocytic thrombospondin-1 is regulated by P2 receptors coupled to protein kinase cascades. Neuroreport 23(12):721–726
Garcia O, Torres M, Helguera P, Coskun P, Busciglio J (2010) A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in Down’s syndrome. PLoS One 5(12):e14200
Lu L, Guo H, Peng Y, Xun G, Liu Y, Xiong Z, Tian D, Liu Y, Li W, Xu X, Zhao J, Hu Z, Xia K (2014) Common and rare variants of the THBS1 gene associated with the risk for autism. Psychiatr Genet 24(6):235–240
Proschel C, Stripay JL, Shih CH, Munger JC, Noble MD (2014) Delayed transplantation of precursor cell-derived astrocytes provides multiple benefits in a rat model of Parkinsons. EMBO Mol Med 6(4):504–518
Reiher FK, Volpert OV, Jimenez B, Crawford SE, Dinney CP, Henkin J, Haviv F, Bouck NP, Campbell SC (2002) Inhibition of tumor growth by systemic treatment with thrombospondin-1 peptide mimetics. Int J Cancer 98(5):682–689
Naganuma H, Satoh E, Asahara T, Amagasaki K, Watanabe A, Satoh H, Kuroda K, Zhang L, Nukui H (2004) Quantification of thrombospondin-1 secretion and expression of alphavbeta3 and alpha3beta1 integrins and syndecan-1 as cell-surface receptors for thrombospondin-1 in malignant glioma cells. J Neurooncol 70(3):309–317
Bogdanov A Jr, Marecos E, Cheng HC, Chandrasekaran L, Krutzsch HC, Roberts DD, Weissleder R (1999) Treatment of experimental brain tumors with trombospondin-1 derived peptides: an in vivo imaging study. Neoplasia 1(5):438–445
Hsu SC, Volpert OV, Steck PA, Mikkelsen T, Polverini PJ, Rao S, Chou P, Bouck NP (1996) Inhibition of angiogenesis in human glioblastomas by chromosome 10 induction of thrombospondin-1. Cancer Res 56(24):5684–5691
Pijuan-Thompson V, Grammer JR, Stewart J, Silverstein RL, Pearce SF, Tuszynski GP, Murphy-Ullrich JE, Gladson CL (1999) Retinoic acid alters the mechanism of attachment of malignant astrocytoma and neuroblastoma cells to thrombospondin-1. Exp Cell Res 249(1):86–101
Harada H, Nakagawa K, Saito M, Kohno S, Nagato S, Furukawa K, Kumon Y, Hamada K, Ohnishi T (2003) Introduction of wild-type p53 enhances thrombospondin-1 expression in human glioma cells. Cancer Lett 191(1):109–119
Anderson JC, Grammer JR, Wang W, Nabors LB, Henkin J, Stewart JE Jr, Gladson CL (2007) ABT-510, a modified type 1 repeat peptide of thrombospondin, inhibits malignant glioma growth in vivo by inhibiting angiogenesis. Cancer Biol Ther 6(3):454–462
Cho S, Kim E (2009) CD36: a multi-modal target for acute stroke therapy. J Neurochem 109(Suppl 1):126–132
Chiquet-Ehrismann R (2004) Tenascins. Int J Biochem Cell Biol 36(6):986–990
Karus M, Denecke B, Wiese S, Faissner A (2011) The extracellular matrix molecule tenascin C modulates expression levels and territories of key patterning genes during spinal cord astrocyte specification. Development 138(24):5321–5331
Pesheva P, Gloor S, Probstmeier R (2001) Tenascin-R as a regulator of CNS glial cell function. Prog Brain Res 132:103–114
Jones EV, Bouvier DS (2014) Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast 2014. doi:10.1155/2014/321209
Garcion E, Faissner A (2001) Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration. Development 128(13):2485–2496
Garwood J, Garcion E, Dobbertin A, Heck N, Calco V Faissner A (2004) The extracellular matrix glycoprotein Tenascin-C is expressed by oligodendrocyte precursor cells and required for the regulation of maturation rate, survival and responsiveness to platelet-derived growth factor. Eur J Neurosci 20(10):2524–2540
Czopka T, Von Holst A, Schmidt G Faissner A (2009) Tenascin C and tenascin R similarly prevent the formation of myelin membranes in a RhoA-dependent manner, but antagonistically regulate the expression of myelin basic protein via a separate pathway. Glia 57(16):1790–1801
Moritz S, Lehmann S, Faissner A, von Holst A (2008) An induction gene trap screen in neural stem cells reveals an instructive function of the niche and identifies the splicing regulator sam68 as a tenascin-C-regulated target gene. Stem Cells 26(9):2321–2331
Faissner A, Kruse J (1990) J1/tenascin is a repulsive substrate for central nervous system neurons. Neuron 5(5):627–637
Lochter A, Vaughan L, Kaplony A, Prochiantz A, Schachner M, Faissner A (1991) J1/tenascin in substrate-bound and soluble form displays contrary effects on neurite outgrowth. J Cell Biol 113(5):1159–1171
Husmann K, Faissner A, Schachner M (1992) Tenascin promotes cerebellar granule cell migration and neurite outgrowth by different domains in the fibronectin type III repeats. J Cell Biol 116(6):1475–1486
Götz B, Scholze A, Clement A, Joester A, Schütte K, Wigger F, Frank R, Spiess E, Ekblom P, Faissner A (1996) Tenascin-C contains distinct adhesive, anti-adhesive, and neurite outgrowth promoting sites for neurons. J Cell Biol 132(4):681–699
Götz M, Bolz J, Joester A, Faissner A (1997) Tenascin-C synthesis and influence on axonal growth during rat cortical development. Eur J Neurosci 9(3):496–506
Meiners S, Geller HM (1997) Long and short splice variants of human tenascin differentially regulate neurite outgrowth. Mol Cell Neurosci 10(1–2):100–116
Meiners S, Powell EM, Geller HM (1999) Neurite outgrowth promotion by the alternatively spliced region of tenascin-C is influenced by cell-type specific binding. Matrix Biol 18(1):75–87
Erickson HP (1993) Tenascin-C, tenascin-R and tenascin-X: a family of talented proteins in search of functions. Curr Opin Cell Biol 5(5):869–876
Wiese S, Karus M, Faissner A (2012) Astrocytes as a source for extracellular matrix molecules and cytokines. Front Pharmacol 3:120
Laywell ED, Dörries U, Bartsch U, Faissner A, Schachner M, Steindler DA (1992) Enhanced expression of the developmentally regulated extracellular matrix molecule tenascin following adult brain injury. Proc Natl Acad Sci U S A 89(7):2634–2638
Hausmann R, Betz P (2000) The time course of the vascular response to human brain injury–an immunohistochemical study. Int J Legal Med 113(5):288–292
Nishio T, Kawaguchi S, Yamamoto M, Iseda T, Kawasaki T, Hase T (2005) Tenascin-C regulates proliferation and migration of cultured astrocytes in a scratch wound assay. Neuroscience 132(1):87–102
Wanner IB, Deik A, Torres M, Rosendahl A, Neary JT, Lemmon VP, Bixby JL (2008) A new in vitro model of the glial scar inhibits axon growth. Glia 56(15):1691–1709. doi:10.1002/glia.20721
Lin HW, Basu A, Druckman C, Cicchese M, Krady JK, Levison SW (2006) Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J Neuroinflammation 30(3):15
Yu YM, Cristofanilli M, Valiveti A, Ma L, Yoo M, Morellini F, Schachner M (2011) The extracellular matrix glycoprotein tenascin-C promotes locomotor recovery after spinal cord injury in adult zebrafish. Neuroscience 183:238–250
Jakovcevski I, Miljkovic D, Schachner M, Andjus PR (2013) Tenascins and inflammation in disorders of the nervous system. Amino Acids 44(4):1115–1127
Zhang Y, Winterbottom JK, Schachner M, Lieberman AR, Anderson PN (1997) Tenascin-C expression and axonal sprouting following injury to the spinal dorsal columns in the adult rat. J Neurosci Res 49(4):433–450
Deller T, Haas CA, Naumann T, Joester A, Faissner A, Frotscher M (1997) Up-regulation of astrocyte-derived tenascin-C correlates with neurite outgrowth in the rat dentate gyrus after unilateral entorhinal cortex lesion. Neuroscience 81(3):829–846
Ikeshima-Kataoka H, Shen JS, Eto Y, Saito S, Yuasa S (2008) Alteration of inflammatory cytokine production in the injured central nervous system of tenascin-deficient mice. In Vivo 22(4):409–413
Gates MA, Fillmore H, Steindler DA (1996) Chondroitin sulfate proteoglycan and tenascin in the wounded adult mouse neostriatum in vitro: dopamine neuron attachment and process outgrowth. J Neurosci 16(24):8005–8018
Soares HD, Potter WZ, Pickering E, Kuhn M, Immermann FW, Shera DM, Ferm M, Dean RA, Simon AJ, Swenson F, Siuciak JA, Kaplow J, Thambisetty M, Zagouras P, Koroshetz WJ, Wan HI, Trojanowski JQ, Shaw LM (2012) Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Biomarkers consortium Alzheimer’s disease plasma proteomics project. Arch Neurol 69(10):1310–1317
Mi Z, Halfter W, Abrahamson EE, Klunk WE, Mathis CA, Mufson EJ, Ikonomovic MD (2016) Tenascin-C Is associated with cored amyloid-β plaques in Alzheimer disease and pathology burdened cognitively normal elderly. J Neuropathol Exp Neurol 75(9):868–876
Xie K, Liu Y, Hao W, Walter S, Penke B, Hartmann T, Schachner M, Fassbender K (2013) Tenascin-C deficiency ameliorates Alzheimer’s disease-related pathology in mice. Neurobiol Aging 34(10):2389–2398
Gutowski NJ, Newcombe J, Cuzner ML (1999) Tenascin-R and C in multiple sclerosis lesions: relevance to extracellular matrix remodelling. Neuropathol Appl Neurobiol 25(3):207–214
Harada M, Kamimura D, Arima Y, Kohsaka H, Nakatsuji Y, Nishida M, Atsumi T, Meng J, Bando H, Singh R, Sabharwal L, Jiang JJ, Kumai N, Miyasaka N, Sakoda S, Yamauchi-Takihara K, Ogura H, Hirano T, Murakami M (2015) Temporal expression of growth factors triggered by epiregulin regulates inflammation development. J Immunol 194(3):1039–1046
Holley JE, Gveric D, Whatmore JL, Gutowski NJ (2005) Tenascin C induces a quiescent phenotype in cultured adult human astrocytes. Glia 52(1):53–58
Zendedel A, Kashani IR, Azimzadeh M, Pasbakhsh P, Omidi N, Golestani A, Beyer C, Clarner T (2016) Regulatory effect of triiodothyronine on brain myelination and astrogliosis after cuprizone-induced demyelination in mice. Metab Brain Dis 31(2):425–433
Niquet J, Jorquera I, Faissner A, Ben-Ari Y, Represa A (1995) Gliosis and axonal sprouting in the hippocampus of epileptic rats are associated with an increase of tenascin-C immunoreactivity. J Neurocytol 24(8):611–624
Nakic M, Mitrovic N, Sperk G, Schachner M (1996) Kainic acid activates transient expression of tenascin-C in the adult rat hippocampus. J Neurosci Res 44(4):355–362
Ferhat L, Chevassus-Au-Louis N, Khrestchatisky M, Ben-Ari Y, Represa A (1996) Seizures induce tenascin-C mRNA expression in neurons. J Neurocytol 25(9):535–546
Mahler M, Ferhat L, Gillian A, Ben-Ari Y, Represa A (1996) Tenascin-C mRNA and tenascin-C protein immunoreactivity increase in astrocytes after activation by bFGF. Cell Adhes Commun 4(3):175–186
Represa A, Ben-Ari Y (1997) Molecular and cellular cascades in seizure-induced neosynapse formation. Adv Neurol 72:25–34
Blümcke I, Beck H, Lie AA, Wiestler OD (1999) Molecular neuropathology of human mesial temporal lobe epilepsy. Epilepsy Res 36(2–3):205–223
Heck N, Garwood J, Loeffler JP, Larmet Y, Faissner A (2004) Differential upregulation of extracellular matrix molecules associated with the appearance of granule cell dispersion and mossy fiber sprouting during epileptogenesis in a murine model of temporal lobe epilepsy. Neuroscience 129(2):309–324
Mercado-Gómez O, Landgrave-Gómez J, Arriaga-Avila V, Nebreda-Corona A, Guevara-Guzmán R (2014) Role of TGF-β signaling pathway on Tenascin C protein upregulation in a pilocarpine seizure model. Epilepsy Res 108(10):1694–1704
Brenneke F, Bukalo O, Dityatev A, Lie AA (2004a) Mice deficient for the extracellular matrix glycoprotein tenascin-r show physiological and structural hallmarks of increased hippocampal excitability, but no increased susceptibility to seizures in the pilocarpine model of epilepsy. Neuroscience 124(4):841–855
Brenneke F, Schachner M, Elger CE, Lie AA (2004b) Up-regulation of the extracellular matrix glycoprotein tenascin-R during axonal reorganization and astrogliosis in the adult rat hippocampus. Epilepsy Res 58(2–3):133–143
Hoffmann K, Sivukhina E, Potschka H, Schachner M, Löscher W, Dityatev A (2009) Retarded kindling progression in mice deficient in the extracellular matrix glycoprotein tenascin-R. Epilepsia 50(4):859–869
Lee Y, Bullard DE, Humphrey PA, Colapinto EV, Friedman HS, Zalutsky MR, Coleman RE, Bigner DD (1988a) Treatment of intracranial human glioma xenografts with 131I-labeled anti-tenascin monoclonal antibody 81C6. Cancer Res 48(10):2904–2910
Lee YS, Bullard DE, Zalutsky MR, Coleman RE, Wikstrand CJ, Friedman HS, Colapinto EV, Bigner DD (1988b) Therapeutic efficacy of antiglioma mesenchymal extracellular matrix 131I-radiolabeled murine monoclonal antibody in a human glioma xenograft model. Cancer Res 48(3):559–566
Higuchi M, Ohnishi T, Arita N, Hiraga S, Hayakawa T (1993) Expression of tenascin in human gliomas: its relation to histological malignancy, tumor dedifferentiation and angiogenesis. Acta Neuropathol 85(5):481–487
Yoshida J, Wakabayashi T, Okamoto S, Kimura S, Washizu K, Kiyosawa K, Mokuno K (1994) Tenascin in cerebrospinal fluid is a useful biomarker for the diagnosis of brain tumour. J Neurol Neurosurg Psychiatry 57(10):1212–1215
Donato G, Lavano A, Volpentesta G, Chirchiglia D, Signorelli CD, Tucci L (1997) Expression of tenascin in astrocytic tumours: too much ado about nothing? J Neurol Neurosurg Psychiatry 63(3):413
Jallo GI, Friedlander DR, Kelly PJ, Wisoff JH, Grumet M, Zagzag D (1997) Tenascin-C expression in the cyst wall and fluid of human brain tumors correlates with angiogenesis. Neurosurgery 41(5):1052–1059
Nie S, Gurrea M, Zhu J, Thakolwiboon S, Heth JA, Muraszko KM, Fan X, Lubman DM (2015) Tenascin-C: a novel candidate marker for cancer stem cells in glioblastoma identified by tissue microarrays. J Proteome Res 14(2):814–822
Sarkar S, Zemp FJ, Senger D, Robbins SM, Yong VW (2015) ADAM-9 is a novel mediator of tenascin-C-stimulated invasiveness of brain tumor-initiating cells. Neuro-oncology 17(8):1095–1105
Vitolo D, Paradiso P, Uccini S, Ruco LP, Baroni CD (1996) Expression of adhesion molecules and extracellular matrix proteins in glioblastomas: relation to angiogenesis and spread. Histopathology 28(6):521–528
Zagzag D, Friedlander DR, Dosik J, Chikramane S, Chan W, Greco MA, Allen JC, Dorovini-Zis K, Grumet M (1996) Tenascin-C expression by angiogenic vessels in human astrocytomas and by human brain endothelial cells in vitro. Cancer Res 56(1):182–189
Castellani P, Siri A, Zardi L, Barbanera A, Dorcaratto A, Viale G (1997) Distribution of tenascin in human malignant gliomas is not related to cell proliferation. J Neurol Neurosurg Psychiatry 62(3):290–301
Hasegawa K, Yoshida T, Matsumoto K, Katsuta K, Waga S, Sakakura T (1997) Differential expression of tenascin-C and tenascin-X in human astrocytomas. Acta Neuropathol 93(5):431–437
Badruddoja MA, Black KL (2006) Improving the delivery of therapeutic agents to CNS neoplasms: a clinical review. Front Biosci 11:1466–1478
Brösicke N, Faissner A (2015) Role of tenascins in the ECM of gliomas. Cell Adh Migr 9(1–2):131–140
Gladson CL (1999) The extracellular matrix of gliomas: modulation of cell function. J Neuropathol Exp Neurol 58(10):1029–1040
Leprini A, Querzé G, Zardi L (1994) Tenascin isoforms: possible targets for diagnosis and therapy of cancer and mechanisms regulating their expression. Perspect Dev Neurobiol 2(1):117–123
Kurpad SN, Zhao XG, Wikstrand CJ, Batra SK, McLendon RE, Bigner DD (1995) Tumor antigens in astrocytic gliomas. Glia 15(3):244–256
Ruoslahti E (1996) Brain extracellular matrix. Glycobiology 6(5):489–492
Zamecnik J (2005) The extracellular space and matrix of gliomas. Acta Neuropathol 110(5):435–442
Kong X, Ma W, Li Y, Wang Y, Guan J, Gao J, Wei J, Yao Y, Lian W, Xu Z, Dou W, Xing B, Ren Z, Su C, Yang Y, Wang R (2015) Does Tenascin have clinical implications in pathological grade of glioma patients?: a systematic meta-analysis. Medicine 94(32):e1330
Hau P, Kunz-Schughart LA, Rümmele P, Arslan F, Dörfelt A, Koch H, Lohmeier A, Hirschmann B, Müller A, Bogdahn U, Bosserhoff AK (2006) Tenascin-C protein is induced by transforming growth factor-beta1 but does not correlate with time to tumor progression in high-grade gliomas. J Neurooncol 77(1):1–7
Xia S, Lal B, Tung B, Wang S, Goodwin CR, Laterra J (2016) Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro-oncology 18(4):507–517
Brack SS, Silacci M, Birchler M, Neri D (2006) Tumor-targeting properties of novel antibodies specific to the large isoform of tenascin-C. Clin Cancer Res 12(10):3200–3208
Herold-Mende C, Mueller MM, Bonsanto MM, Schmitt HP, Kunze S, Steiner HH (2002) Clinical impact and functional aspects of tenascin-C expression during glioma progression. Int J Cancer 98(3):362–369
Mai J, Sameni M, Mikkelsen T, Sloane BF (2002) Degradation of extracellular matrix protein tenascin-C by cathepsin B: an interaction involved in the progression of gliomas. Biol Chem 383(9):1407–1413
Maiuri F, Cappabianca P, Gangemi M, De Caro Mdel B, Esposito F, Pettinato G, de Divitiis O, Mignogna C, Strazzullo V, de Divitiis E (2006) Clinical progression and familial occurrence of cerebral cavernous angiomas: the role of angiogenic and growth factors. Neurosurg Focus 21(1):e3
Taga T, Suzuki A, Gonzalez-Gomez I, Gilles FH, Stins M, Shimada H, Barsky L, Weinberg KI, Laug WE (2002) alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer 98(5):690-
Viale G, Dorcaratto A, Castellani P, Zardi L (2002) Tenascin-C in astrocytic tumors. Surg Neurol 57(4):286
Bigner DD, Brown MT, Friedman AH, Coleman RE, Akabani G, Friedman HS, Thorstad WL, McLendon RE, Bigner SH, Zhao XG, Pegram CN, Wikstrand CJ, Herndon JE 2nd, Vick NA, Paleologos N, Cokgor I, Provenzale JM, Zalutsky MR (1998) Iodine-131-labeled antitenascin monoclonal antibody 81C6 treatment of patients with recurrent malignant gliomas: phase I trial results. J Clin Oncol 16(6):2202–2212
Hirata E, Arakawa Y, Shirahata M, Yamaguchi M, Kishi Y, Okada T, Takahashi JA, Matsuda M, Hashimoto N (2009) Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci 100(8):1451–1459
Marriott CJ, Thorstad W, Akabani G, Brown MT, McLendon RE, Hanson MW, Coleman RE (1998) Locally increased uptake of fluorine-18-fluorodeoxyglucose after intracavitary administration of iodine-131-labeled antibody for primary brain tumors. J Nucl Med 39(8):1376–1380
Onishi M, Ichikawa T, Kurozumi K, Date I (2011) Angiogenesis and invasion in glioma. Brain Tumor Pathol 28(1):13–24
Sarkar S, Nuttall RK, Liu S, Edwards DR, Yong VW (2006) Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res 66(24):11771–11780
Sarkar S, Yong VW (2010) Reduction of protein kinase C delta attenuates tenascin-C stimulated glioma invasion in three-dimensional matrix. Carcinogenesis 31(2):311–317
Silacci M, Brack SS, Späth N, Buck A, Hillinger S, Arni S, Weder W, Zardi L, Neri D (2006) Human monoclonal antibodies to domain C of tenascin-C selectively target solid tumors in vivo. Protein Eng Des Sel 19(10):471–478
Zalutsky MR, Archer GE, Garg PK, Batra SK, Bigner DD (1996) Chimeric anti-tenascin antibody 81C6: increased tumor localization compared with its murine parent. Nucl Med Biol 23(4):449–458
Zukiel R, Nowak S, Wyszko E, Rolle K, Gawronska I, Barciszewska MZ, Barciszewski J (2006) Suppression of human brain tumor with interference RNA specific for tenascin-C. Cancer Biol Ther 5(8):1002–1007
Riva P, Arista A, Franceschi G, Frattarelli M, Sturiale C, Riva N, Casi M, Rossitti R (1995) Local treatment of malignant gliomas by direct infusion of specific monoclonal antibodies labeled with 131I: comparison of the results obtained in recurrent and newly diagnosed tumors. Cancer Res 55(23 Suppl):5952s–5956s
Vincent AJ, Lau PW, Roskams AJ (2008) SPARC is expressed by macroglia and microglia in the developing and mature nervous system. Dev Dyn 237(5):1449–1462
Mendis DB, Malaval L, Brown IR (1995) SPARC, an extracellular matrix glycoprotein containing the follistatin module, is expressed by astrocytes in synaptic enriched regions of the adult brain. Brain Res 676(1):69–79
Mendis DB, Brown IR (1994) Expression of the gene encoding the extracellular matrix glycoprotein SPARC in the developing and adult mouse brain. Brain Res Mol Brain Res 24(1–4):11–19
Mendis DB, Ivy GO, Brown IR (1996) SC1, a brain extracellular matrix glycoprotein related to SPARC and follistatin, is expressed by rat cerebellar astrocytes following injury and during development. Brain Res 730(1–2):95–106
Murphy-Ullrich JE (2001) The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest 107(7):785–790
Brekken RA, Sage EH (2001) SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol 19(8):816–827
Yan Q, Sage EH (1999) SPARC, a matricellular glycoprotein with important biological functions. J Histochem Cytochem 47(12):1495–1506
Ikemoto M, Takita M, Imamura T, Inoue K (2000) Increased sensitivity to the stimulant effects of morphine conferred by anti-adhesive glycoprotein SPARC in amygdala. Nat Med 6(8):910–915
Kupprion C, Motamed K, Sage EH (1998) SPARC (BM-40, osteonectin) inhibits the mitogenic effect of vascular endothelial growth factor on microvascular endothelial cells. J Biol Chem 273(45):29635–29640
Vafadar-Isfahani B, Ball G, Coveney C, Lemetre C, Boocock D, Minthon L, Hansson O, Miles AK, Janciauskiene SM, Warden D, Smith AD, Wilcock G, Kalsheker N, Rees R, Matharoo-Ball B, Morgan K (2012) Identification of SPARC-like 1 protein as part of a biomarker panel for Alzheimer’s disease in cerebrospinal fluid. J Alzheimers Dis 28(3):625–636
Richens JL, Vere KA, Light RA, Soria D, Garibaldi J, Smith AD, Warden D, Wilcock G, Bajaj N, Morgan K, O’Shea P (2014) Practical detection of a definitive biomarker panel for Alzheimer’s disease; comparisons between matched plasma and cerebrospinal fluid. Int J Mol Epidemiol Genet 5(2):53–70
Lloyd-Burton SM, York EM, Anwar MA, Vincent AJ, Roskams AJ (2013) SPARC regulates microgliosis and functional recovery following cortical ischemia. J Neurosci 33(10):4468–4481
Baumann E, Preston E, Slinn J, Stanimirovic D (2009) Post-ischemic hypothermia attenuates loss of the vascular basement membrane proteins, agrin and SPARC, and the blood–brain barrier disruption after global cerebral ischemia. Brain Res 1269:185–197
Mendis DB, Ivy GO, Brown IR (2000) Induction of SC1 mRNA encoding a brain extracellular matrix glycoprotein related to SPARC following lesioning of the adult rat forebrain. Neurochem Res 25(12):1637–1644
Turtoi A, Musmeci D, Naccarato AG, Scatena C, Ortenzi V, Kiss R, Murtas D, Patsos G, Mazzucchelli G, De Pauw E, Bevilacqua G, Castronovo V (2012) Sparc-like protein 1 is a new marker of human glioma progression. J Proteome Res 11(10):5011–5021
Schultz C, Lemke N, Ge S, Golembieski WA, Rempel SA (2002) Secreted protein acidic and rich in cysteine promotes glioma invasion and delays tumor growth in vivo. Cancer Res 62(21):6270–6277
Golembieski WA, Thomas SL, Schultz CR, Yunker CK, McClung HM, Lemke N, Cazacu S, Barker T, Sage EH, Brodie C, Rempel SA (2008) HSP27 mediates SPARC-induced changes in glioma morphology, migration, and invasion. Glia 56(10):1061–1075
Thomas SL, Alam R, Lemke N, Schultz LR, Gutiérrez JA, Rempel SA (2010) PTEN augments SPARC suppression of proliferation and inhibits SPARC-induced migration by suppressing SHC-RAF-ERK and AKT signaling. Neuro-oncology 12(9):941–955
Thomas SL, Schultz CR, Mouzon E, Golembieski WA, El Naili R, Radakrishnan A, Lemke N, Poisson LM, Gutiérrez JA, Cottingham S, Rempel SA (2015) Loss of sparc in p53-null astrocytes promotes macrophage activation and phagocytosis resulting in decreased tumor size and tumor cell survival. Brain Pathol 25(4):391–400
McClung HM, Golembieski WA, Schultz CR, Jankowski M, Schultz LR, Rempel SA (2012) Deletion of the SPARC acidic domain or EGF-like module reduces SPARC-induced migration and signaling through p38 MAPK/HSP27 in glioma. Carcinogenesis 33(2):275–284
Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, Rich JN (2007) Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene 26(28):4084–4094
Yunker CK, Golembieski W, Lemke N, Schultz CR, Cazacu S, Brodie C, Rempel SA (2008) SPARC-induced increase in glioma matrix and decrease in vascularity are associated with reduced VEGF expression and secretion. Int J Cancer 122(12):2735–2743
Liu H, Xu Y, Chen Y, Zhang H, Fan S, Feng S, Liu F (2011) RNA interference against SPARC promotes the growth of U-87MG human malignant glioma cells. Oncol Lett 2(5):985–990
Capper D, Mittelbronn M, Goeppert B, Meyermann R, Schittenhelm J (2010) Secreted protein, acidic and rich in cysteine (SPARC) expression in astrocytic tumour cells negatively correlates with proliferation, while vascular SPARC expression is associated with patient survival. Neuropathol Appl Neurobiol 36(3):183–197
Seno T, Harada H, Kohno S, Teraoka M, Inoue A, Ohnishi T (2009) Downregulation of SPARC expression inhibits cell migration and invasion in malignant gliomas. Int J Oncol 34(3):707–715
Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA 108(32):E440–E449
Jones EV, Bernardinelli Y, Tse YC, Chierzi S, Wong TP, Murai KK (2011) Astrocytes control glutamate receptor levels at developing synapses through SPARC-beta-integrin interactions. J Neurosci 31(11):4154–4165
Au E, Richter MW, Vincent AJ, Tetzlaff W, Aebersold R, Sage EH, Roskams AJ (2007) SPARC from olfactory ensheathing cells stimulates Schwann cells to promote neurite outgrowth and enhances spinal cord repair. J Neurosci 27(27):7208–7221
Lorber B, Chew DJ, Hauck SM, Chong RS, Fawcett JW, Martin KR (2015) Retinal glia promote dorsal root ganglion axon regeneration. PLoS One 10(3):e0115996
Filmus J, Capurro M, Rast J (2008) Glypicans. Genome Biol 9(5):224
Svensson G, Awad W, Håkansson M, Mani K, Logan DT (2012) Crystal structure of N-glycosylated human glypican-1 core protein: structure of two loops evolutionarily conserved in vertebrate glypican-1. J Biol Chem 287(17):14040–14051
David G, Lories V, Decock B, Marynen P, Cassiman JJ, Van den Berghe H (1990) Molecular cloning of a phosphatidylinositol-anchored membrane heparan sulfate proteoglycan from human lung fibroblasts. J Cell Biol 111(6 Pt 2):3165–3176
Karthikeyan L, Maurel P, Rauch U, Margolis RK, Margolis RU (1992) Cloning of a major heparan sulfate proteoglycan from brain and identification as the rat form of glypican. Biochem Biophys Res Commun 188(1):395–401
Karthikeyan L, Flad M, Engel M, Meyer-Puttlitz B, Margolis RU, Margolis RK (1994) Immunocytochemical and in situ hybridization studies of the heparan sulfate proteoglycan, glypican, in nervous tissue. J Cell Sci 107(Pt 11):3213–3222
Ivins JK, Litwack ED, Kumbasar A, Stipp CS, Lander AD (1997) Cerebroglycan, a developmentally regulated cell-surface heparan sulfate proteoglycan, is expressed on developing axons and growth cones. Dev Biol 184(2):320–332
Veugelers M, Vermeesch J, Reekmans G, Steinfeld R, Marynen P, David G (1997) Characterization of glypican-5 and chromosomal localization of human GPC5, a new member of the glypican gene family. Genomics 40(1):24–30
Saunders S, Paine-Saunders S, Lander AD (1997) Expression of the cell surface proteoglycan glypican-5 is developmentally regulated in kidney, limb, and brain. Dev Biol 190(1):78–93
Bandtlow CE, Zimmermann DR (2000) Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev 80(4):1267–1290
Wang W, Dow KE (1997) Differential regulation of neuronal proteoglycans by activation of excitatory amino acid receptors. Neuroreport 8(3):659–663
Akita K, Toda M, Hosoki Y, Inoue M, Fushiki S, Oohira A, Okayama M, Yamashina I, Nakada H (2004) Heparan sulphate proteoglycans interact with neurocan and promote neurite outgrowth from cerebellar granule cells. Biochem J 383(Pt 1):129–138
Rawson JM, Dimitroff B, Johnson KG, Rawson JM, Ge X, Van Vactor D, Selleck SB (2005) The heparan sulfate proteoglycans Dally-like and Syndecan have distinct functions in axon guidance and visual-system assembly in Drosophila. Curr Biol 15(9):833–838
Luxardi G, Galli A, Forlani S, Lawson K, Maina F, Dono R (2007) Glypicans are differentially expressed during patterning and neurogenesis of early mouse brain. Biochem Biophys Res Commun 352(1):55–60
Qiao D, Yang X, Meyer K, Friedl A (2008) Glypican-1 regulates anaphase promoting complex/cyclosome substrates and cell cycle progression in endothelial cells. Mol Biol Cell 19(7):2789–2801
Jen YH, Musacchio M, Lander AD (2009) Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Dev 4:33. doi:10.1186/1749-8104-4-33
van Horssen J, Otte-Höller I, David G, Maat-Schieman ML, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM (2001) Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer’s disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol 102(6):604–614
van Horssen J, Kleinnijenhuis J, Maass CN, Rensink AA, Otte-Höller I, David G, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM (2002) Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol Aging 23(4):537–545
Watanabe N, Araki W, Chui DH, Makifuchi T, Ihara Y, Tabira T (2004) Glypican-1 as an Abeta binding HSPG in the human brain: its localization in DIG domains and possible roles in the pathogenesis of Alzheimer’s disease. FASEB J 18(9):1013–1015
Verbeek MM, Otte-Höller I, van den Born J, van den Heuvel LP, David G, Wesseling P, de Waal RM (1999) Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer’s disease brain. Am J Pathol 155(6):2115–2125
Williamson TG, Mok SS, Henry A, Cappai R, Lander AD, Nurcombe V, Beyreuther K, Masters CL, Small DH (1996) Secreted glypican binds to the amyloid precursor protein of Alzheimer’s disease (APP) and inhibits APP-induced neurite outgrowth. J Biol Chem 271(49):31215–31221
Cappai R, Cheng F, Ciccotosto GD, Needham BE, Masters CL, Multhaup G, Fransson LA, Mani K (2005) The amyloid precursor protein (APP) of Alzheimer disease and its paralog, APLP2, modulate the Cu/Zn-Nitric Oxide-catalyzed degradation of glypican-1 heparan sulfate in vivo. J Biol Chem 280(14):13913–13920
O’Callaghan P, Sandwall E, Li JP, Yu H, Ravid R, Guan ZZ, van Kuppevelt TH, Nilsson LN, Ingelsson M, Hyman BT, Kalimo H, Lindahl U, Lannfelt L, Zhang X (2008) Heparan sulfate accumulation with Abeta deposits in Alzheimer’s disease and Tg2576 mice is contributed by glial cells. Brain Pathol 18(4):548–561
Timmer NM, van Horssen J, Otte-Holler I, Wilhelmus MM, David G, van Beers J, de Waal RM, Verbeek MM (2009) Amyloid beta induces cellular relocalization and production of agrin and glypican-1. Brain Res 1260:38–46
Qiao D, Meyer K, Mundhenke C, Drew SA, Friedl A (2003) Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells. Specific role for glypican-1 in glioma angiogenesis. J Biol Chem 278(18):16045–16053
Qiao D, Meyer K, Friedl A (2013) Glypican 1 stimulates S phase entry and DNA replication in human glioma cells and normal astrocytes. Mol Cell Biol 33(22):4408–4421
Hill JJ, Jin K, Mao XO, Xie L, Greenberg DA (2012) Intracerebral chondroitinase ABC and heparan sulfate proteoglycan glypican improve outcome from chronic stroke in rats. Proc Natl Acad Sci USA 109(23):9155–9160
Fico A, de Chevigny A, Melon C, Bohic M, Kerkerian-Le Goff L, Maina F, Dono R, Cremer H (2014) Reducing glypican-4 in ES cells improves recovery in a rat model of Parkinson’s disease by increasing the production of dopaminergic neurons and decreasing teratoma formation. J Neurosci 34(24):8318–8323
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486(7403):410–414
de Wit J, O’Sullivan ML, Savas JN, Condomitti G, Caccese MC, Vennekens KM, Yates JR 3rd, Ghosh A (2013) Unbiased discovery of glypican as a receptor for LRRTM4 in regulating excitatory synapse development. Neuron 79(4):696–711
Ko JS, Pramanik G, Um JW, Shim JS, Lee D, Kim KH, Chung GY, Condomitti G, Kim HM, Kim H, de Wit J, Park KS, Tabuchi K, Ko J (2015) PTPσ functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. Proc Natl Acad Sci USA 112(6):1874–1879
Li J, Ye L, Owen S, Weeks HP, Zhang Z, Jiang WG (2015) Emerging role of CCN family proteins in tumorigenesis and cancer metastasis (Review). Int J Mol Med 36(6):1451–1463
Ishida J, Kurozumi K, Ichikawa T, Otani Y, Onishi M, Fujii K, Shimazu Y, Oka T, Shimizu T, Date I (2015) Evaluation of extracellular matrix protein CCN1 as a prognostic factor for glioblastoma. Brain Tumor Pathol 32(4):245–252
Albrecht C, von Der Kammer H, Mayhaus M, Klaudiny J, Schweizer M, Nitsch RM (2000) Muscarinic acetylcholine receptors induce the expression of the immediate early growth regulatory gene CYR61. J Biol Chem 275(37):28929–28936
Malik AR, Liszewska E, Jaworski J (2015) Matricellular proteins of the Cyr61/CTGF/NOV (CCN) family and the nervous system. Front Cell Neurosci 9:237
Kondo Y, Nakanishi T, Takigawa M, Ogawa N (1999) Immunohistochemical localization of connective tissue growth factor in the rat central nervous system. Brain Res 834(1–2):146–151
Le Dréau G, Kular L, Nicot AB, Calmel C, Melik-Parsadaniantz S, Kitabgi P, Laurent M, Martinerie C (2010) NOV/CCN3 upregulates CCL2 and CXCL1 expression in astrocytes through beta1 and beta5 integrins. Glia 58(12):1510–1521
Guo J, Cheng C, Chen CS, Xing X, Xu G, Feng J, Qin X (2016) Overexpression of Fibulin-5 attenuates ischemia/reperfusion injury after middle cerebral artery occlusion in rats. Mol Neurobiol 53(5):3154–3167
Guadall A, Orriols M, Rodríguez-Calvo R, Calvayrac O, Crespo J, Aledo R, Martínez-González J, Rodríguez C (2011) Fibulin-5 is up-regulated by hypoxia in endothelial cells through a hypoxia-inducible factor-1 (HIF-1α)-dependent mechanism. J Biol Chem 286(9):7093–7103
Yanagisawa H, Schluterman MK, Brekken RA (2009) Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal 3(3–4):337–347
Walther M, Kuklinski S, Pesheva P, Guntinas-Lichius O, Angelov DN, Neiss WF, Asou H, Probstmeier R (2000) Galectin-3 is upregulated in microglial cells in response to ischemic brain lesions, but not to facial nerve axotomy. J Neurosci Res 61(4):430–435
Hu L, Dong MX, Zhao H, Xu GH, Qin XY (2016) Fibulin-5: a novel biomarker for evaluating severity and predicting prognosis in patients with acute intracerebral haemorrhage. Eur J Neurol 23(7):1195–1201
Sirko S, Irmler M, Gascón S, Bek S, Schneider S, Dimou L, Obermann J, De Souza Paiva D, Poirier F, Beckers J, Hauck SM, Barde YA, Götz M (2015) Astrocyte reactivity after brain injury-: the role of galectins 1 and 3. Glia 63(12):2340–2361
Qu WS, Wang YH, Ma JF, Tian DS, Zhang Q, Pan DJ, Yu ZY, Xie MJ, Wang JP, Wang W (2011) Galectin-1 attenuates astrogliosis-associated injuries and improves recovery of rats following focal cerebral ischemia. J Neurochem 116(2):217–226
Venkatesan C, Chrzaszcz M, Choi N, Wainwright MS (2010) Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J Neuroinflammation 7:32
Endo T (2005) Glycans and glycan-binding proteins in brain: galectin-1-induced expression of neurotrophic factors in astrocytes. Curr Drug Targets 6(4):427–436
Imbe H, Okamoto K, Kadoya T, Horie H, Senba E (2003) Galectin-1 is involved in the potentiation of neuropathic pain in the dorsal horn. Brain Res 993(1–2):72–83
McGraw J, Oschipok LW, Liu J, Hiebert GW, Mak CF, Horie H, Kadoya T, Steeves JD, Ramer MS, Tetzlaff W (2004) Galectin-1 expression correlates with the regenerative potential of rubrospinal and spinal motoneurons. Neuroscience 128(4):713–719
Acknowledgements
This work was supported by a Merit Review from the US Department of Veterans Affairs and by a National Institutes of Health grant (DK063311).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jayakumar, A.R., Apeksha, A. & Norenberg, M.D. Role of Matricellular Proteins in Disorders of the Central Nervous System. Neurochem Res 42, 858–875 (2017). https://doi.org/10.1007/s11064-016-2088-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2088-5